
Abstract
Obesity is nowadays considered a pandemic which prevalence’s has been steadily increasingly in western countries. It is a dynamic, complex, and multifactorial disease which propitiates the development of several metabolic and cardiovascular diseases, as well as cancer. Excessive adipose tissue has been causally related to cancer progression and is a preventable risk factor for overall and cancer-specific survival, associated with poor prognosis in cancer patients. The onset of obesity features a state of chronic low-grade inflammation and secretion of a diversity of adipocyte-derived molecules (adipokines, cytokines, hormones), responsible for altering the metabolic, inflammatory, and immune landscape. The crosstalk between adipocytes and tumor cells fuels the tumor microenvironment with pro-inflammatory factors, promoting tissue injury, mutagenesis, invasion, and metastasis. Although classically established as a risk factor for cancer and treatment toxicity, recent evidence suggests mild obesity is related to better outcomes, with obese cancer patients showing better responses to treatment when compared to lean cancer patients. This phenomenon is termed obesity paradox and has been reported in different types and stages of cancer. The mechanisms underlying this paradoxical relationship between obesity and cancer are still not fully described but point to systemic alterations in metabolic fitness and modulation of the tumor microenvironment by obesity-associated molecules. Obesity impacts the response to cancer treatments, such as chemotherapy and immunotherapy, and has been reported as having a positive association with immune checkpoint therapy. In this review, we discuss obesity’s association to inflammation and cancer, also highlighting potential physiological and biological mechanisms underlying this association, hoping to clarify the existence and impact of obesity paradox in cancer development and treatment.
Similar content being viewed by others

Introduction
Although extensively researched, obesity and cancer are both pathologies exhibiting steadily-increasing and fast-paced expansion worldwide. In 2015, 2 billion people (27% of the world population) was considered overweight [1]. Alongside, cancer incidence also increased in recent years. In 2018, there were 18.1 million new cancer cases worldwide [2]. These ongoing epidemics accounted for over 4 million and 9.6 million deaths, respectively, amounting for billions of dollars in costs related to treatment and hospitalization [3,4,5]. This great toll presents major challenges to healthcare systems worldwide. Western countries, in particular, show increasing prevalence of obesity and obesity-related morbidities [6]. Regarding cancer, a similar situation can be described. Although some types of cancer showed a decrease in death rates in the last 20 years—due to better understanding and treatment of the disease, cancer cases are on the rise [2, 7, 8]. A 60% increase in the number of cancer cases is expected in the next 20 years [9]. In low- and middle-income countries, this increase could reach 81%, as developmental and economic factors also influence the expansion of both diseases [10,11,12,13].
The onset of obesity is linked to increased occurrence of cancer (one of many obesity-associated diseases) and a more aggressive, metastatic phenotype of tumor cells [14, 15]. This link is usually defined in terms of the systemic low-grade chronic obesity-induced inflammation, considered a hallmark of cancer establishment and progression [16, 17]. Changes in adipose tissue composition, function, and secretion have a direct impact on tumor microenvironment (TME) and the immune system [18, 19]. Both weight loss and the transdifferentiation between different types of adipose tissue (white, brown, and beige) are considered viable strategies to prevent cancer incidence. While the white adipose tissue (WAT) secretes a plethora of inflammatory mediators (adipokines, cytokines, hormones), inducing tissue damage/remodeling and propitiating cancer progression, the beige and brown adipose tissues (BAT) are responsible for adaptive energy expenditure, inducing thermogenesis and protecting against obesity and, therefore, obesity-related cancer [20, 21]. The functioning of cancer cells and the organization of the TME are affected not only by adipocyte dysfunction but also by immune cells residing in the adipose tissue microenvironment (ATM), such as macrophages and lymphocytes [22].
Although obesity is classically considered a pro-carcinogenic condition, in some cases, overweight and/or obesity seemingly induces a protective status against certain stages and types of cancer, since it may boost anti-tumor immunotherapy. This conflictual idea is termed obesity paradox and entails improved outcomes of overweight and class 1 obesity (Body Mass Index-BMI = 25–34.9 kg/m2) cancer patients when compared to lean, although this effect is not observed in all patients and cancers [23, 24]. As for the reasons why this takes place, a few hypotheses, both methodological and epidemiological, come to mind, such as biases in the use of BMI as a reliable measure of obesity and body composition, the use of excessive adipose tissue as an energy reserve (improving response to treatment), and better absorption, metabolism, and response to treatment in obese patients [25,26,27]. Also, loss of adipose and muscle tissue (cachexia and sarcopenia) is associated with higher toxicity and mortality of cancer patients, suggesting mild obesity could compensate for this loss, actually turning the accumulation of adipose tissue into a protective tool against cancer development and/or progression [28, 29]. It is important to highlight obesity is still considered a risk factor for most types of cancer, even non-obesity-associated tumors and tumors in which prevalence is not related to obesity. Severe obesity does not confer the same advantages, which raises questions about which mechanisms take part pro- and anti-tumoral properties of adipose tissue.
The mechanisms and pathways ruling this co-regulation between adipose tissue and the TME are yet to be fully described, but evidence points to the involvement of the immune system’s inflammatory and cellular response, through mediators exchanged between the adipose tissue, immune cells, and cancer cells [30]. Considering the variety of adipocytes, cancer cells and immune cells in the TME, tumorigenesis and tumor progression are targeted by cells (or secretion from these cells) with diverse functioning and morphology, acting antagonistically or synergically, depending on a wide range of factors (number, activation, polarization, localization) [31, 32].
Given the metabolic, inflammatory, and immune abnormalities of obese cancer patients, together with the complex crosstalk between adipocytes and cancer cells, and its implications in cancer treatment, this review focuses on debating the implications of obesity and the obesity paradox in cancer development and immunotherapy response, as well as diving into the physiological and biological mechanisms underlying this paradox.
Obesity and inflammation modulation
Obesity and metabolic syndrome: global health burdens
A direct consequence of lifestyle changes, the statistics regarding obesity are escalating worldwide, impacting more than 6 hundred thousand people in 2016. Although the definition and classification of obesity are controversial, patients displaying a BMI equal or higher than 30 kg/m2 are considered as presenting the obese phenotype (WHO, 2020). Obesity is intimately related to elevated risk of developing a large set of pathologies as it associates with chronic low-grade inflammation in many tissues, as adipose tissue (AT), liver, skeletal muscle, intestine, pancreatic islets, and brain [33,34,35,36]. Its inflammatory characteristics cope obesity with type 2 diabetes (T2D), heart disorders, non-alcoholic fatty liver diseases (NAFLDs), and certain malignancies, conditions which elevated morbidity favor the increased death rate characteristic of the obese state [37, 38].
The most frequent component of metabolic syndrome (MS) [39], abdominal obesity is characterized to be a waist circumference of more than 102 cm in men and more than 88 cm in women [40]. Elevated amount of visceral AT (VAT) relates to increased soluble tumor necrosis factor-alpha (TNF-α) receptor 2 [41], incident hypertension [42], and adverse metabolic risk profile [43]. Although the criteria for defining and diagnosing MS has changed throughout the years [44], this lifestyle disease drastically increases the risk of cardiovascular and cerebrovascular disorders [45] and associates with insulin resistance (IR) [46].
Subcellular disturbances and oxidative stress
Several studies connect adipocyte subcellular alterations with the inflammatory and metabolic derangements observed in MS-affected individuals, including disrupted mitochondrial and endoplasmic reticulum (ER) functions [47, 48]. Excessive food intake favors reactive oxygen species (ROS) production and mitochondrial dynamic dysfunction [49]. Mitochondria, the powerhouse of the cell, oxidize glucose and free fatty acids (FFAs) to produce adenosine triphosphate (ATP) [50]. However, this small organelle is a relevant ROS source inside the cell [51]: nutrient excess in adipocytes leads to augmented electron supply to the mitochondria electron transport chain (ETC) [52], favoring ROS generation. ROS damages mitochondrial constituents [53], impacting on cell viability [54]. Mitochondrial dysfunction contribute to VAT inflammation through the ROS and mitochondrial-derived damage-associated molecular patterns (DAMPs) release [55].
Nutrient overload is also implicated in endoplasmic reticulum (ER) stress [56]. The largest organelle in the cell, ER can be found at the cell endoplasm [57] and is the main actor in protein production, calcium (Ca2+) storage, and lipid biosynthesis [58]. When proteins accumulate in ER lumen, the homeostatic mechanism unfolded protein response (UPR) is initiated [59], leading to ROS formation and ER stress [60]. ER stress influence VAT inflammatory status through inducing TNFα and IL-6 expression, activating JNK pathway [61], and favoring nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) signaling [62].
Adipocyte cell death and AT inflammation
An evidence that diets rich in saturated lipids directly influence MS onset is that progressive lipid accumulation in adipocytes correlates with cell hypertrophy and enlarged VAT, which cause adipocyte stress and cytokine secretion [63]. The inflammatory and oxidative microenvironment of VAT impairs healthy adipose expansion [64]. Although adipocytes secrete several inflammatory molecules [65, 66], most mediators are produced by immune cells present in VAT of obese animals and humans [34]. During metabolic syndrome, these inflammatory cells secrete massive amounts of proinflammatory cytokines, as Interferon γ (IFNγ), Interleukin 6 (IL-6), and TNFα [35, 67, 68], lipid mediators, as Leukotriene B4 (LTB4) [69], and chemokines, as Monocyte Chemoattractant Protein-1 (MCP1) [70,71,72]. Stressed adipocytes present deregulated adipokine secretory pattern, secreting more proinflammatory adipokines, as leptin, and less of the anti-inflammatory adiponectin [73]. The key players in VAT chronic inflammation are decreased Th2 cells [74] and Treg [75] and increased neutrophils [76], mast cells [77], innate lymphoid cells (ILCs) [78, 79], CD8- [80] and CD4-positive T cells (Th1 and Th17) [81], B cells (B2) [82], and macrophages (M1-like) [83, 84].
T and B cells and macrophages form crown-like structures (CLSs) around dead or dying adipocytes [80, 85, 86], which display apoptotic and pyroptotic cell death signaling [87, 88]. Differently from apoptosis [89], pyroptosis depends on inflammasome activity for membrane pore formation and secretion of IL-18 and IL-1β [90, 91]. NLR family pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2) inflammasomes are molecular pattern sensors [92] that, through processing caspase-1, enable cytokine secretion and maturation of Gasdermin D (GSDMD), the cell death executor [90]. Membrane integrity loss leads to extravasation of intracellular contents including HMGB1 [93, 94], an alarming that, in conjunction with other intracellular factors, as ATP [95], amplifies VAT inflammation [88].
The rapid VAT enlargement and the increased VAT oxygen consumption that cause MS favors the occurrence of hypoxia, which enhance adipocyte MCP-1 and LTB4 secretion [96] and increase macrophage M1-like polarization [97]. A MS component [98], elevated fasting glucose leads to enhanced advanced glycation end products (AGEs) and its receptor (RAGE), which induce AIM2 inflammasome activation [99, 100]. In addition, persistent hyperglycemia in rodent models modulates gut immune function and microbiota [101]. Also highlighting the impact of diet on MS development, high-fat diet (HFD) mouse models display exacerbated VAT inflammation [102] due to the action of saturated FFAs on NLRP3 [103], and Toll-like receptors-2 (TLR-2) and 4 (TLR-4) [104]. Microbiota is also altered by HFD, favoring increased lipopolysaccharide (LPS)-containing bacteria and augmented intestinal permeability [105], resulting in metabolic endotoxemia and systemic inflammation worsening [106], implicating dysbiosis in MS pathology [107].
AT inflammation and insulin resistance
IR strongly correlates with MS [108]. IR was first observed by Himsworth et al., who could verify that individuals display discrepant blood glycemia after insulin infusion [109]. Many studies connect IR with oxidative stress and chronic low-grade inflammation [110, 111]. Increased VAT levels of proinflammatory mediators, including MCP-1 [112], IFN γ [80, 113], TNF [65, 83], and IL-6 [114] induce insulin resistance (IR) in VAT. In addition, studies evaluating the absence of neutrophils [76], mast cells [77], ILCs [79, 115], CD8- [80] and CD4-positive T cells (Th1 and Th17) [113], B cells (B2) [82], and macrophages (M1-like) [65, 112] inform improved VAT IR. M1-like migration to VAT was shown to precede IR in HFD mouse model [116]. The anti-inflammatory adiponectin, downregulated in subjects with obesity and T2D [117], is also an insulin sensitizer [118]. In addition to the effect of caspase-1 on IR through modulating inflammatory status, this protein also influences VAT metabolic function [119]. Caspase-1 and inflammasome activity influence the gut microbiota as well [120], and human and mouse experimental endotoxemia studies indicate circulating LPS as an IR inducer [121].
Inflammation exacerbate IR and vice versa [36, 122]. IR is associated with many detrimental systemic effects [122]. As IR impair proper glucose uptake by skeletal muscle and favor liver gluconeogenesis, hyperglycemia leads to pancreatic cytotoxicity and β-cell death [36, 123]. Furthermore, IR induces VAT hormone sensitive lipase activity and lipolysis, which increase the levels of circulating FFA [37], and the effects of inflammation on preadipocyte/adipocyte favor ectopic fat deposition in organs as liver and skeletal muscle, leading to systemic IR [36, 123]. Considering their detrimental effects on VAT physiology here described, oxidative stress, chronic low-grade inflammation, and IR are considered to drive metabolic syndrome, enhancing the risk for several life-threatening diseases [45].
Therefore, exacerbated food intake and sedentary lifestyle leads to mitochondrial dysfunction and ER stress, subcellular disturbances connected to VAT oxidative stress and inflammation. The disrupted inflammatory status in VAT favors immune cells infiltration, proliferation, and polarization to proinflammatory profiles. VAT dysfunction and the stressful microenvironment induce adipocyte apoptotic and pyroptotic cell death, which further exacerbate inflammation. Dietary factors influence systemic and VAT inflammatory status through impacting on immune cells and on microbiota, the latter being key on regulating intestinal barrier integrity and, during dysbiosis, cope with elevated circulating LPS level (Fig. 1). Inflammatory status is intimately related to insulin sensitivity through several molecular pathways, and IR and inflammation, exacerbating each other, act as drivers of the elevated morbidity, mortality, and financial burden of metabolic syndrome.

Obesity-related physiological states, immune response and cancer development. Comparison between underweight/lean, overweight/mild obesity and obese phenotypes, regarding their physiological states, immune responses and cancer development. While both extremes—underweight and obese—show poor survival and disadvantages regarding cancer development and progression, mild obesity, in overweight patients, has been described as a protective phenotype, allowing for improved immunotherapy response and survival of cancer patients
Obesity, cancer, and the obesity paradox
Cancer: definition and pathology statistics
Cancer, rather than a single disease, is a set of over 100 pathologies which share a common denominator: excessive proliferation. The transformation of normal cells into malignant cells is a largely studied, but still not fully understood process with multiple causes, a different perspective of the previous genetic-driven transformation [124]. The recently established—and continuously updated—hallmarks of cancer, comprise multiple characteristics and capacities which, when acquired or expressed by cells, allow for sustained proliferation, resistance to cell death, immortality, and enable tumoral cells to grow and spread throughout the organism, ultimately enabling the emergence of malignant tumors, i.e., cancer [125].
In 2020, in the United States alone, 1,806,590 diagnosed cancer cases are estimated (around 4950 new cases/day). Prostate, breast, lung, colon, and skin make up roughly half of the sites of tumor development. Regarding cancer-related deaths, it is estimated that 606,520 individuals perished from cancer in 2020. Even though in the last 20 years or so the cancer incidence rate for men and women has stabilized (due to better understanding, diagnosis, and screening), cancer is still the second leading cause of deaths in the United States. Risk factors for the development of cancer include not only genetic predisposition, but also the exposure to a gamut of environmental and behavioral factors, such as excessive alcohol consumption, cigarette smoking, infections, and, as discussed here, obesity [126, 127].
Obesity and cancer: overview of a complex relationship
According to data from the American Cancer Society, in 2014, 7.8% (122,536) of all cancers in the United States and 6.5% (38,188) of all cancer deaths were attributed to overweight or obesity [128]. Globally, estimations point to 481,000 new cancer cases being related to obesity, designating excessive body adiposity as a well-established risk factor for cancer development [129]. These cases, however, are not equally distributed between all cancer types and countries. In men, two thirds of these cases were from kidney and colon cancer, while, in women, approximately three quarters were from postmenopausal breast, corpus uteri and colon cancer [130, 131]. As Whiteman and Wilson pointed, the United States had the highest fraction of colorectal, pancreatic, and postmenopausal breast cancer cases attributable to overweight and obesity [132]. Added to the fact that, also in the United States, there has been a fast-paced growth in overweight and obesity rates, with 66% of adults and 33% of children being considered overweight or obese, the landscape is nothing short of alarming [133]. This trend seems to also be true to developing countries, such as Brazil, where the aforementioned cancers account for just about half of all cancer cases identified in the country by the year 2012 [134].
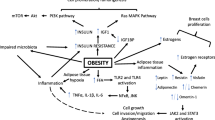
A variety of different types of cancer have already been described as having an increased risk when associated to excessive body fatness, such as cancers from the gastrointestinal tract (esophagus, cardia, stomach, pancreas, gallbladder, liver, colon, rectum), breast (postmenopausal, specifically ER+ breast cancer risk, whereas ER− or TNBC is little or inversely associated with obesity), kidney, thyroid, prostate, ovary, endometrium, multiple myeloma, and meningioma [135, 136]. There is significant evidence of positive associations between obesity and cancer [129]. Excessive body fatness is tied to systemic and tumor microenvironmental inflammation, usually reported as a chronic low-grade pro-inflammatory state, altering the immune response, insulin resistance, insulin-like growth factors, and sex hormones pathways, inducing specific lipids, and secreting various adipokines and inflammatory cytokines [23, 137,138,139]. In league, these obesity-related processes account for a thriving environment for tumor initiation, development, and progression [136, 140, 141] (Fig. 2).

Biological mechanisms of obesity in cancer development and progression. Pathophysiological mechanisms regarding obesity effects upon cancer development and progression. The secretion of adipokines and release of free fatty acids by adipocytes induces several different systemic and cellular responses. The combination of metabolic alterations induced by the adipose tissue results in the activation of several signaling pathways responsible for cellular growth, proliferation, invasion, migration, inhibition of apoptosis, angiogenesis and induction of inflammation, all of which are hallmarks of cancer development
Seemingly paradoxical reports for other cancers, however, indicate that overweight (BMI ≥ 25 kg/m2 and obesity (BMI ≥ 30 kg/m2) are related to more favorable long-term survival post-surgery or treatment. Patients showed increased long-term, recurrence-free, and overall survival, while displaying reduced morbidities in different cancer types, such as renal cell carcinoma and non-small cell lung cancer (NSCLC), suggesting high BMI as an independent predictor for better cancer survival [28, 142,143,144,145].
The obesity paradox
In face of such opposing reports, studies termed the occurrence of the obesity paradox, a different relation between obesity as a risk factor for diseases, including cancer, in which higher adiposity (primarily determined using the BMI index) is associated to better survival, response to treatment, and better clinical outcomes, different from the commonplace where a higher BMI index would aid disease development or point to reduced chances of survival [28, 146].
The obesity paradox was first described in diseases and conditions other than cancer, with supporting literature regarding cardiovascular disease (CVD), atherosclerosis, kidney failure, and T2D [147,148,149,150,151]. Review of clinical and epidemiological literature shows some types of cancer also fit the obesity paradox. In patients with lung, bladder, breast cancer (premenopausal), and melanoma, to name a few, overweight/obese patients displayed better long-term survival rate, reduced overall morbidity, and reduced in-hospital mortality [142, 152].
These studies endorse the complexity of obesity and its effects upon the metabolism, considering that, for some types of cancer, the obese phenotype predicts for better clinical outcomes and long-term prognosis. Zhang et al. (2017) regarded the obesity paradox as an under-explored scientific fact, rather than an unexpected paradox—a point of view shared by this review. The collective evidence supports both a deleterious and a protective role for obesity in cancer, reflected in its definition as a risk factor for cancer development and as a protective phenotype for established cancers. A common factor in most conditions in which the obesity paradox is observed is the perpetuation of inflammation. The lack of inflammation resolution is central to the development of chronic diseases and tumoral growth and expansion [147]. By fostering the development of cancer, inflammation and immune mediators may be pieces of the puzzle for explaining the cellular and molecular basis of the obesity paradox. Even so, this line of research is still in need of further exploration [125, 147, 153].
There is a lot of debate about the obesity paradox and potential issues with previous studies in which it was described. Namely, aspects such as confounding variables (smoking, age and other comorbidities), reverse causation (weight change as a consequence rather than a cause of cancer), differences in attentive and medical care (control of hypertension and prevention of hyperglycemia in obese patients), collider stratification biases and the fact that BMI (the most preponderant measure for obesity) does not differentiate between subcutaneous (SAT), visceral (VAT), and other types of adipose tissue [23, 28, 136, 142, 154]. When examining previous meta-analyses, attention is also given for the timing of BMI measurement, if pre- or post-diagnosis, treatment, or surgery [136, 155].These are usually considered limitations in research concerning the obesity paradox, for they may partly explain the observed differences in obesity’s effects upon each type of cancer. Since many extensive reviews take part on describing the aspects for why this paradox may or may not be true, other than an illusionary causality [23, 136], we focus on the physiological states and biological mechanisms which possibly underlie these observations, also commenting upon the impacts of adiposity over the immune system and immunotherapy, in an effort to provide biological basis for the results and evidence present in the literature.
Obesity and cancer-related physiological states
Healthy versus unhealthy obesity
Obesity is recognized as an adverse factor in the outset of a wide gamut of diseases, metabolic disorders, and several types of cancer [156]. It is the main cause of CVD, cancer mortality and all-cause mortality [157,158,159,160]. Notwithstanding, a proportion of obese individuals might benefit from the obesity-related phenotype and show decreased risk for obesity-related metabolic complications, despite having a high BMI [161, 162]. This has been credited to differences in body fat distribution and heterogeneous cardiometabolic profiles, originating the ‘metabolically healthy obese’ (MHO) subgroup [157, 163], comprising obese individuals (BMI > 30 kg/m2) with reduced risk of cardiovascular morbidity and mortality [156]. This subgroup is classified as obese, even though individuals do not have MS and show appropriate metabolic profile [157]. Populational frequencies range from 10 to 50%, with recent reports of an overall prevalence of 35% among 40 populations of obese adults [164, 165].
Studies comparing different obese subgroups and considering the stratification by presence or absence of MS show distinct rates for total mortality [166, 167]. In general, MHO individuals have lower risk of cardiovascular and all-cause mortality when compared to metabolically unhealthy obese (MUO) and metabolically unhealthy non-obese (MUNO) individuals [168]. In certain populations, these differences are also observed when comparing metabolically healthy obese (MHO) to metabolically healthy non-obese (MHNO) individuals, indicating metabolic capability has an overall stronger impact upon mortality than BMI alone, since poor metabolic health status contributes more to mortality than high BMI indexes [169, 170]. Besides adipose tissue distribution and insulin resistance, metabolic risk factors encompass lipid profiling, blood pressure, inflammation, and physical fitness [156].
When discussing the physiological foundation and biological mechanisms ruling metabolic health obesity—as well as cancer development-, genetically modified mouse models of metabolically healthy obesity are a useful tool. Leptin-deficient mice, or those overexpressing the mitochondrial outer membrane protein (mitoNEET), or adipose-specific glucose transporter type 4, show increased non-visceral adipose tissue deposition, expansion of subcutaneous adipose tissue, reduced fat content in the liver and a metabolically beneficial adipocytokine secretion pattern [171, 172]. Immune-wise, these mice exhibit reduced infiltration of macrophages in adipose tissue, and low systemic inflammation. A plausible explanation is that higher subcutaneous adipose content, supported by adipose tissue de novo lipogenesis, in conjunction with reduced inflammatory signaling, enable a healthier phenotype [173,174,175,176]. Concurrently, in humans, observations about the MHO phenotype are similar to those of animal models. Metabolic healthy obese individuals present reduced fat depots in the skeletal muscle and liver fat deposition (related to insulin sensitivity), while also displaying expansion of subcutaneous adipose tissue [177, 178].
Together, improved metabolic health and better control over inflammation, MHO patients (despite a higher BMI), show better prognosis against cancer development and cancer-related adversities even when compared to lean (metabolically unhealthy) patients. In conjunction, they promote tolerance to cancer therapy and post-surgery complications, resulting in improved long-term survival [24, 28]. Previous studies shed a light on the incidence rates of gastric cancer in MHO patients. Compared to all other subtypes, the incidence and hazard ratios of gastric cancer were lower than all other groups [179]. Similarly, the risks of colorectal neoplasia in MHO individuals are lower when compared to their non-obese counterparts, whether healthy or unhealthy [180]. The same is true to bladder cancer [181].
When MHO subjects are compared to lean controls, this advantage may not always be observed, or even be inversed back to the usual obesity-as-a-risk-factor scenario. A 20 year follow-up showed no difference in mortality and CV morbidity risks when comparing MHO to non-obese subjects [182]. When discussing cancer—or the obesity paradox, for that matter, it is not unusual for studies to contradict each other. Not all cancer types behave similarly, and confirming this, the MHO phenotype was linked to increased incidence of gastroesophageal reflux disease and erosive esophagitis (risk factors for esophageal adenocarcinoma) when compared to other metabolically healthy groups [183, 184]. A large scale (12,542,390 participants) comparison between MHO and MHNO individuals for incidence of any type of cancer showed obesity’s effects outrange metabolic health and increase the risk of developing cancer [185].
In the face of the divergent studies about metabolically health obesity, and its fairly recent definition, there are limitations for considering it an advantage, at least concerning cancer development and pathology. Not only metabolic health status is a transient phenotype, but there is a lack of uniformity in the outcome of studies concerning cancer and a few other diseases [180,181,182,183, 186]. Obesity is not, in most situations, a safe condition which would justify lack of treatment or consideration. Even without metabolic dysfunction, therapeutic weight loss is still instructed to obese individuals [185]. Nevertheless, metabolically health obesity can serve as a model for mechanistic studies regarding obesity and obesity-associated diseases. Also, in the clinical scenario, it might assist in a more personalized and risk-stratified obesity treatment, since obesity is taken into consideration when defining therapeutic procedures and predicting patient’s prognosis of cancer [28, 187].
Meet me halfway: too lean is also not good
Another physiological state that needs to be thoroughly considered in the context of cancer and body weight, is cachexia. Cachexia’s exact definition is still debated, but it is regarded as a metabolic syndrome associated to disease, a systemic inflammatory response marked by loss of muscle mass (sarcopenia), associated or not to loss of fat mass, resulting in functional decline because of a negative protein-energy balance [188, 189]. It is often cited as a paraneoplastic syndrome, i.e., symptoms resulting from tissue damage outside the tumor site, due to the crosstalk between the tumor and the immune system [190]. Cachexia is related to 20% of cancer-related deaths and displays high prevalence in pancreatic and gastric cancer (87%), as well as colon, lung, prostate (61%) and breast (40%) cancer. Expectedly, it is an indicator of poor prognosis and increased mortality in cancer patients [191,192,193]. The unbalance in nutritional homeostasis is mediated by tumor-induced alterations in the metabolism, impairing the capacity to drive-off infections and resist the adverse effects of chemotherapy and radiotherapy. Cachexia also reduces the efficacy of anti-cancer treatments [191, 194]. Besides cancer, cachexia is also associated with an array of acute and chronic inflammatory conditions [195,196,197].
In the context of obesity, obesity-associated cancer, and the obesity paradox, overweight and obese subjects would have larger nutritional reserves than lean subjects, better dealing with the adverse effects of cachexia and better resisting surgical interventions [142]. This implies that patients with higher BMI are less vulnerable to cachexia-induced effects, displaying a more favorable outcome [28]. Considering cachexia and sarcopenia account for reductions in BMI, having a higher BMI indicates a tendency for better prognosis, as described in studies comparing obese, underweight, and lean patients with lung cancer and metastatic melanoma [193, 198, 199]. However, there is a catch. Obesity is very heterogeneous, and the ratio between fat and muscle mass is highly variable between individuals [200]. A know phenomenon which can be overlooked in patients with high BMI is sarcopenic obesity, in which excessive adiposity is accompanied by sarcopenia [201, 202]. Indeed, computed tomography (CT) scans reveal that the conventional criteria of assessing obesity using BMI is not precise enough for the detection of cachexia and sarcopenia, regardless of overall body weight [203]. In patients with breast cancer, postdiagnosis weight gain is often associated with loss of muscle mass, rendering BMI alone as an ineffective tool to assess sarcopenic obesity [204].
Accumulated evidence shows sarcopenic obesity as a predictor of morbidity and mortality in cancer, emphasizing the importance of lean mass maintenance in survival. Cachexia and sarcopenia are related to poor outcome and increased chemotherapy-induced toxicity in patients with lung cancer. In pancreatic cancer, they predict survival and worst postoperative outcome. In solid tumors from the respiratory and gastrointestinal tract, cachexia indicates poorer functional status [189, 205, 206].
Another important hallmark of cancer cachexia is systemic inflammation and an increase in pro-inflammatory cytokines, fueling cancer progression [188, 207, 208]. In cachexia, WAT persistently produces and secretes pro-inflammatory mediators, contributing to the onset and persistence of inflammation [209, 210]. Increased expression of TNF-α, IL-1β and IL-10 was observed in WAT adipocytes from different regions [211]. During cancer-associated cachexia, SAT could impact early cancer development, secreting higher numbers of inflammatory cytokines when compared to the mesenteric adipose tissue [212]. Regulation of inflammation in the adipocytes is carried on by NF‐κB and inflammasome pathways activation [209, 211]. Indeed, in cachectic cancer patients, there is increased expression of NF-κBp65 and its target genes [209]. NLRP3 inflammasome and caspase-1 pathways are also activated, given the secretion of IL-1β [213,214,215]. Knockout animal models for NLRP3 and caspase-1 are resistant to the development of obesity and show enhanced insulin sensitivity when compared to controls [211, 216, 217].
Cancer and cachexia are also accompanied of adipose atrophy (mainly loss of WAT), through increased adipocyte lipolysis, reduced lipid deposition, and decreased adipogenesis. In cancer patients, these factors have been linked to reduced quality of life and shorter survival, regardless of BMI [28, 218]. Remarkably, in animal models of cancer cachectic mice, browning of WAT precedes skeletal muscle atrophy [219]. It is suggested that inhibiting the switch from white to brown adipose tissue prevents severe loss of muscle mass and that alterations in the adipose tissue metabolism are a prerequisite for skeletal muscle atrophy [220, 221]. Indeed, blocking adipocyte lipolysis and browning seems to protect mice from lung carcinoma-induced cachexia, improving survival [222, 223]. The exact mechanisms that mediate the crosstalk between loss of fat mas to skeletal muscle atrophy are still not clear [224]. It has been proposed that inflammation, induction of lipases and reduced AMP-activated protein kinase activity contribute to tumor-induced lipolysis [225, 226]. By storing a larger quantity of adipose tissue, obese patients could have an advantage when dealing with the metabolic demand and wasting conditions of cancer and cachexia, preventing both adipose and muscular tissue loss, or at least better resisting these processes, which makes sense partly explaining the protective role of excessive adiposity (i.e., obesity paradox) in cancer and cachexia [28, 227].
Home is where good fat is
When diagnosing adiposity, BMI is still predominant, disregarding its inability to differentiate lean mass from adipose tissue, and/or the different types and locations of adipose tissue, which possess distinguishable functioning and reverberations upon metabolism and cancer progression [228]. Accordingly, obesity-induced metabolic abnormalities are often associated to adipose tissue location, rather than total amount of adipose tissue [229, 230]. In obesity, VAT and SAT, which have distinct morphology and function, are of major interest [231, 232]. 5–20% of total body fat is constituted of VAT, while SAT accounts for approximately 80% [233]. SAT is often described as benign (or mildly related to disease development), insulin-sensitive, less lipolytic and more lipogenic, while VAT, is related to elevated lipid turnover, reduced plasticity (transdifferentiation), higher vascularization, and is usually infiltrated with immune cells (macrophages and lymphocytes) [232, 234, 235].
VAT is considered an endocrine organ, responsible for synthesizing several molecules which regulate appetite, innate and adaptive immunity, hematopoiesis, and angiogenesis, i.e., processes involved in physiologic and homeostatic maintenance, but also pathologic development. VAT is a known causal factor of insulin resistance, hypertension, CVD, and cancer [98, 236,237,238]. VAT secretes proinflammatory mediators, growth factors, hormones (such as estrogen), and adipokines, all of which contribute to the establishment and progression of diseases [237, 239]. Up to now, over 15 adipokines have been related to cancer progression, a number which is expected to grow with the advance of research in this area [240, 241].
Synergistically, FFAs resulting from VAT lipolysis act upon the liver, inducing insulin resistance and altering glucose metabolism. FFAs stimulate mutagenic pathways and contribute to proliferation, growth, and migration of cancer cells [231, 233]. FFAs modulate gene expression, activate the mTOR/PI3K pathway, reduce the expression of inhibitors of cell proliferation and promote metastasis [242,243,244]. The proximity of VAT to vital organs (liver, heart, and colon) contributes to its role in inducing pathophysiological alterations such as metabolic syndrome and cancer [245, 246]. In contrast, large depots of SAT are associated with better prognosis, reduced mortality risk and better overall survival in gastrointestinal, respiratory, kidney and prostate cancer [247, 248].
Considering the extensive distinction between subsets of adipose tissue, in the clinic, it would be recommended to measure body fat and adipose tissue distribution using more accurate parameters, other than BMI. Waist circumference, waist-to-hip ratio, and skinfold thickness are more strongly associated with visceral fat and cancer risk than BMI, even though evidence is conflicting, since these methods define both VAT and SAT at the waist level [138, 153, 249]. Other techniques, such as dual-energy X-ray absorptiometry, bioelectrical impedance analysis, and computed tomography would be ideal for VAT and SAT quantification, except its application may not be feasible for large populational studies [250, 251].
Biological mechanisms of obesity in cancer: how fat does what it does
Adipocytes and the adipose tissue are described as dynamic and metabolically active organs, capable of secreting a variety of molecules, with both local and systemic repercussions [252, 253]. Secretion from adipocytes comprehends hormones, cytokines and adipokines, which exert autocrine, paracrine, and endocrine signaling upon many organs [230]. Many mechanisms linking obesity to cancer risk and mortality are suggested, such as insulin resistance, hyperinsulinemia, hyperglycemia, oxidative stress, inflammation and/or adipokine production [254]. Epidemiological evidence about the association of circulating adipokines and cancer include, but are not limited to, breast, gastric, colon, endometrium, kidney, prostate, and pancreatic cancer [240, 246, 255, 256]. In conjunction with obesity-driven chronic inflammation, adipocyte dysfunction plays a fundamental role in adiposity-induced tumorigenesis [31]. This association is clear in cancers developing in or nearby adipocyte-rich environments (particularly breast cancer), and those which invade fat-rich sites, like ovarian and gastric cancers [257,258,259].
Adipokines: the communicators of adipose tissue
Adipokines (also called adipocytokines), compose a diverse group of over 20 different hormones and signaling molecules derived from adipocytes [240]. Adipokines are responsible for regulating various physiological processes, including energy balance, lipid metabolism, glucose homeostasis, insulin resistance/sensitivity, angiogenesis, and inflammation. In obesity, the excess of fat tissue results in adipocyte dysfunction and promotes adipose tissue-related disorders (adiposopathies) [254]. Adipocytokines are also implicated in carcinogenesis, tumor progression, recurrence, and metastasis. Between all adipokines, two are of particular importance in obesity and cancer, and will be more thoroughly discussed: leptin and adiponectin. Assuredly, other adipokines have considerable consequences for obesity-related disorders and cancer progression. More comprehensive reviews of the multiple adipokines can be found in the reviews by Saygin et al., and Gallo et al. [240, 260].
Leptin: a hunger for cancer development
Leptin, the appetite suppressant hormone, was identified in 1994 as the driving factor for obesity in the ob/ob mice model. This mutation caused early-onset obesity and metabolic alterations, and identification of leptin was essential for consolidating the adipose tissue as an endocrine tissue with effects even upon the brain. The only previously described adipocytokine was a cytokine, TNF-α, in 1993 [261,262,263]. Leptin has been described as a potent proinflammatory stimulatory hormone on human peripheral blood monocytes [264] and an enhancer of the activation and proliferation of human circulating Th1 lymphocytes [265]. Indeed, impaired cell-mediated immunity is observed in mice with a defect in leptin (ob/ob) or its receptor (db/db) [266,267,268], and both leptin and leptin receptor are possible targets for intervention in the immunometabolic mediated pathophysiology [269]. Circulating leptin concentrations are closely related to obesity, where higher levels of this hormone are observed in obese individuals [270, 271]. Likely, decreased levels of leptin have been demonstrated in severe malnourished infants [272], whereas an increase in leptin and the immunological recovery was observed after refeeding of malnourished infants [273]. Leptin is classically described as regulator of food intake and energy expenditure, where elevated circulating levels of leptin are associated with adipose tissue inflammation and implicated in breast, colon, prostate, pancreas, ovary, and lung cancers [254, 260, 274,275,276].
In fact, leptin has a dual role in cancer [277]. Leptin is associated with tumor growth and promotion for its mitogenic, antiapoptotic, pro-angiogenic, and pro-inflammatory effects, while also promoting invasion and migration [240, 254, 278]. The intracellular pathways involved in leptin signaling include JAK/STAT, ERK, and PI3K, culminating in increased proliferation, survival, invasion, and angiogenesis, all of which are hallmarks of cancer development [240]. Leptin can also modulate cell death by inhibiting apoptosis through the upregulation of Bcl expression (namely Bcl-xl, Bak, and Bax) and induce angiogenesis by stimulating HIF-1α and NF-kB, resulting in increased production of vascular endothelial growth factor (VEGF) in breast cancer models. In the MCF-7 cell line, leptin can decrease p53 expression, favoring cancer survival [279, 280]. Mice models of obesity also highlight the role of leptin in survival and maintenance of cancer stem cells, complemented by its regulation of NANOG, SOX2, and OCT4 [281, 282]. Leptin also increases invasiveness, inhibits mitochondrial respiration, and blocks endoplasmic reticulum stress signaling (inhibiting apoptosis) in gastric, colorectal, and liver cancer, respectively [256, 283].
When comparing prostate cancer patients (including advanced stages) to patients with benign prostate hyperplasia or early stage prostate cancer, leptin expression was increased, indicating leptin expression can be used as a biomarker for prostate cancer staging and prognosis [284, 285]. Furthermore, leptin expression is associated to with chemotherapy resistance in gastroesophageal adenocarcinomas, indicating leptin antagonists may be useful in the development of new therapeutic alternatives [286].
Adiponectin: just like leptin, but you want it
In the context of obesity, another major adipokine is adiponectin, a regulator of glucose homeostasis and fatty acid oxidation, also implied in insulin resistance and diabetes [287]. Rather than leptin, circulating levels of adiponectin are inversely related to adiposity and body fat mass [260, 288]. Obesity, particularly increased visceral fat (also associated with increased oxidative stress and increased inflammation), is accompanied of decreased adiponectin [289]. Accordingly, reduced levels of adiponectin are also a feature observed in different types of cancer and a more aggressive and advanced stage of cancer progression, as observed in breast, colon, esophagus, liver, and endometrial cancer [290, 291]. Indeed, a systematic review and meta-analysis recently showed increased adiponectin was significantly associated with decreased risk of cancer [292]. Adiponectin not only affects cancer cells by directly inhibiting proliferation and invasion, but also indirectly, by reducing insulin resistance and insulin levels [293].
AdipoR1 and AdipoR2 receptors are the best examined receptors that bind to adiponectin. These receptors activate 5′-adenosine monophosphate activated protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPAR)-α pathways, increasing energy expenditure and fatty acid oxidation, while also improving insulin sensitivity [294, 295]. Concurrently, activation of AMPK inhibits the PI3K/Akt/mTOR pathway, increasing expression of p53 and Bax, decreasing DNA replication and translation of cell cycle and angiogenesis genes. This sequence of events causes decreased cell growth and proliferation. Furthermore, low expression of adiponectin receptors (observed in obesity) is associated to endothelial dysfunction, higher histological grade, myometrial invasion, and lymph node metastasis [296, 297]. Taking this into account, adiponectin has opposite effects to leptin in cancer, promoting apoptosis and reducing proliferation, migration, and inflammation [240]. Epidemiological studies agree with the proposed mechanisms, showing low levels of adiponectin are associated to increased number, stage, and risk of endometrial, prostate, esophageal and colorectal cancer [298,299,300,301].
Inflammation: fuel it, and cancer likes it
Given the first described adipokine was TNF-α, a cytokine, the effects of obesity on inflammation, specifically the production of inflammatory cytokines is not an accident. Obesity is extensively described as a state of chronic low-grade systemic inflammation and is an established contributor for cancer progression, fueling inflammation and cancer development by producing inflammatory mediators able to affect the tumor microenvironment [302, 303]. The expansion of adipocytes numbers and area brings with it the apoptosis of these cells. Dying adipocytes are surrounded by monocytes and macrophages, giving rise to crown-like structures, a hallmark of adipose inflammation [304]. The close contact and interaction between adipocytes and immune cells propitiate enhanced production of multiple inflammatory factors, including cytokines, pro-inflammatory adipokines, while also enhancing lipolysis and liberation of FFAs [302, 305]. A growing number of studies has described interactions with adipocytes, such as lymphocytes, eosinophils, neutrophils, mast cells and foam cells [306]. Although there is a lack of knowledge about the initiation of this interaction, macrophage polarization, infiltration of neutrophils, and reduction/dysfunction/exhaustion of T cells are observed and suggested as inducers of adipose inflammation, strengthening the link between obesity and inflammation [307,308,309].
Alterations in the production and secretion of adipokines can also impact the production of inflammatory factors. Considering the obese phenotype, reduced circulating adiponectin is implied in the production of pro-inflammatory cytokines, such as TNF-α and IL-6, while increased leptin stimulates the production of IL-1, IL-6, IL-12, TNF-α, plasminogen activator inhibitor-1 (PAI-1), leukotriene B4 (LTB4) and cyclooxygenase 2 (COX2) [237, 310]. These cytokines are secreted by adipocytes, cancer-associated adipocytes (CAA), macrophages and other immune cells present in the site of inflammation or tumorigenesis [257, 311, 312]. This cytokine cascade creates an ideal tumor microenvironment that facilitates the acquisition of more aggressive and invasive phenotypes [310].
Adipocytes are a major source of TNF-α, secreted in response to FFAs derived from lipolysis (via JNK signaling pathway). TNF-α, on its own, (via ERK signaling pathway) induces lipolysis and, consequently, the release of FFAs. This molecular talk between adipocytes and macrophages gives rise to a positive feedback mechanism, a paracrine loop comprising FFAs and TNF-α that exacerbates inflammation, contributing to a chronic inflammatory dysfunction in the adipose tissue [313, 314]. It is worth to note that TNF-α also contributes to insulin resistance in overall obesity, altering the metabolic landscape [315]. Concurrently, IL-1β and IL-6 expressions are increased. Both cytokines are extensively associated with insulin resistance, tumorigenesis, and cancer progression [316,317,318]. The presence of adipose tissue and CAAs in the TME (particularly in the fat-rich breast cancer) may aggravate cancer progression and provide the tumor with adipose-derived stem cells, which contribute to cancer development and angiogenesis [302]. Moreover, cancer cells may alter the neighboring adipocytes’ phenotypes, promoting lipolysis, altering the release of adipokines, secreting matrix metalloproteinases, and producing reactive oxygen species in response to FFAs, which, in turn promotes extensive tissue damage and fuels the TME even more [319, 320].
Besides adipokines and inflammation, other processes and pathways are involved in the relationship between obesity and cancer progression. Alterations in insulin-like growth factors’ pathways, induction of hypoxia and HIF-1α signaling, induction of ER stress, alterations in estrogens levels, and modulation of microbiota have all been cited as significant modulators of the effects of obesity in cancer [230, 237, 260].
Obesity and the obesity paradox in cancer treatment
Obesity paradox in traditional cancer treatment: the more you try, the less it fits
Obesity and obesity-related disorders have major implications in cancer development and management. Regarding the existence of an obesity paradox, for a few specific types of cancer, obesity can be seen as a phenotypic advantage. Contradicting the evidence of obesity as a risk factor for most cancer types, the obesity paradox has been regarded as an existent, but not largely determinant feature of obesity in cancer. In fact, studies continue to suggest that multimodal interventions, adding body composition, physical fitness, and nutritional and metabolic enhancement of obese individuals to therapeutic intervention, is key for cancer management and treatment [28, 311]. Correspondingly, traditional, well-stablished therapeutic strategies against cancer (chemotherapy, radiotherapy, and surgery) are modulated by the host’s adiposity, with different outcomes and behaviors in the face of treatment. The obesity paradox can be observed in response to cancer treatment, but, as always, evidence is conflicting depending on cancer type, obesity parameters, and treatment [24, 321].
Surgical outcomes in obesity-associated cancer demonstrate both the inexistence of obesity-conferred advantages of high BMI, and the association of high BMI with increased surgical complications in breast, gastric, colorectal, hepatic, and pancreatic cancer [322,323,324,325]. Only a handful of cancer types (such as such as gastric, pancreatic and lung cancer) associate obesity to lower mortality and increased long-term survival [326,327,328]. Notwithstanding, as previously stated, literature discrepancies, methodological biases, and obesity-induced alterations in physiologic states (i.e., cachexia, sarcopenia, insulin resistance/sensitivity) may partly explain these results. Along the same lines, radiotherapy shows inferior outcomes when associated to obesity in breast, prostate, and cervical cancer [329,330,331,332].
Chemotherapy, in general, follows the same road, showing reduced efficacy in obese cancer patients, with few available reports linking the obesity paradox to chemotherapy efficiency. On one hand, higher BMI was associated with worse response to doxorubicin and increased hematologic toxicity in breast cancer. On the other hand, obese patients with lung cancer showed no association between treatment with carboplatin and survival/toxicity, indicating obesity may not be detrimental in some types of cancer [28, 333,334,335]. A recent analysis of patients with lung cancer described decreases in BMI during chemotherapy were associated with poor survival, which would fit the obesity paradox, since higher BMI would be a protective tool [205]. Some studies also show obese patients with less myelosuppression and toxic effects while undergoing chemotherapy, particularly in lung, breast, and gynecologic cancers [336,337,338,339]. These findings, however, are debatable since many studies describe obesity as a major factor in promoting cancer resistance to chemotherapy [334, 340,341,342].
Poor response to chemotherapy in obesity: just not getting enough?
When reviewing obesity-induced effects on chemotherapy, however, attention must be given to a common practice in the clinic, called dose capping. Dose capping chemotherapy means that obese patients with greater body surface receive lower, or sub-optimal doses of anti-cancer drugs. These inappropriate calculations arise from the use of ideal body weight—instead of the patients’ own body weight—for dose determination [24, 26, 321, 343]. This may be a well-intentioned practice, concerned about excessive toxicity and possible comorbidities in obese patients. However, reduced dosage is associated with reduced survival, and increased cancer recurrence and mortality [344]. Apart from scarce evidence of a positive relation between obesity and chemotherapy efficiency, the underlying biological mechanisms are not yet clear. It was proposed that excessive adipose tissue can dilute drugs, alter blood pressure, and blood flow (affecting drug distribution), and result in liver fat deposition (affecting drug clearance) [28, 336, 345, 346].
When discussing classic, well-established therapeutic strategies against cancer, once again, the obesity paradox is not a rule, but sheds a light upon new mechanistic pathways linking obesity and cancer development. Recent literature still points to one more instance in which obesity seemingly presents beneficial effects: cancer immunotherapy.
Immunotherapy, cancer, and obesity
Cancer immunotherapy: stopping cancer’s escape plan
Immunity against cancer involves complex interactions between tumor cells, the host, and the TME, in a process called cancer immunoediting [347]. The understanding of the interplays between the tumor, immune components, and the intrinsic capacity of individuals to fight the tumor are leading to discoveries and adoption of strategies using immunological mechanisms to combat the most diverse types of tumors, as well as the possibility of predicting an individual’s response to immunotherapy [348,349,350].
Cancer immunotherapy is a therapeutic approach that uses certain components of the body’s immune system to recognize, control and combat cancer cells with greater specificity. There are several strategies that are used to increase immunity against tumor cells, which represent a change in the cancer treatment paradigm [348, 349]. Some of the new immunotherapeutic tools are based on the adoptive transfer of T cells, dendritic cells or Natural Killer (NK) cells, oncolytic viruses, biological modifiers such as vaccines and cytokines, monoclonal antibodies against specific antigens or immunological checkpoint inhibitors, such as inhibitors of cytotoxic T lymphocyte associated protein 4 (CTLA-4), T cell programmed death receptor 1 (PD-1) or one of its ligands (PDL-1 or PDL-2) [350,351,352,353,354,355,356,357,358].
Many new antigens (neoantigens) associated with tumors can be expressed as mutated proto-oncogenes, expressed in excess, or aberrantly expressed. These so-called tumors associated antigens are antigens that are not naturally expressed in these cell types and are presented via molecules of the major histocompatibility complex (MHC). The recognition of an MHC/peptide complex (pMHC) by a T cell antigen receptor (TCR) is insufficient for complete activation of T lymphocytes. Complete T cell activation requires a second co-stimulatory signal, in which receptors and ligands on the surface of professional antigen presenting cells (APC) stimulate T lymphocyte activation, differentiation, proliferation and function. There are several receptor/ligand combinations between T cells and APCs which can induce stimulatory signals, such as the coupling between CD28 and CD80/86, or inhibitory signals, such as PD-1 and PD-L1/PD-L2. These inhibitory signals are called immune checkpoints [349].
Immune checkpoint molecules, like CTLA-4, PD-1, lymphocyte-activation gene 3 (LAG3), T-cell immunoglobulin and mucin-domain containg-3 (TIM-3), and V-domain Ig suppressor of T cell activation (VISTA), among others, are important negative regulators of immune system activation and are generally expressed by APCs and regulatory T cells to inhibit cytotoxic T cells and preserve tolerance [359]. The inhibition of T cells leads to the suppression of the activation of these cells, inducing a state of anergy, with decreased proliferation, effector functions, and production of cytokines. The checkpoint molecules can also be expressed in high and sustained levels by CD4+ or CD8+ T cells activated in response to factors derived from the TME, indicating a state of cellular exhaustion. The ligands for these inhibitory receptors can be found in tumor cells and several myeloid cells [359,360,361]. Cancer cells are capable of molding the TME, inhibiting T cells by engaging with immunological checkpoints, and recruiting non-tumor cells such as fibroblasts, macrophages, and endothelial cells, amongst others. These components of the TME help and protect tumors from immune system recognition and elimination, and, thus, circumvent the tumor immunovigilance (or immunosurveillance), resulting in the escape of tumor cells from the immune system response [347, 349, 359, 362].
Tumor cells, however, can also develop a different interrelation with the immune system. Immunoselective pressure drives a process called immunoediting, in which tumor cells shape the antigenic repertoire of their subclones, eliminating the more immunogenic variants. Tumor cells can further subvert infiltrating immune cells, using them to promote their growth, tumor progression and inducing an immunosuppressive microenvironment [347, 363, 364]. Polarization of immune cells leading to a pro-inflammatory cytokine profile, or infiltration of innate and adaptive immune cells in the tumor may contribute to tumor progression, instead of killing. Reportedly, macrophages, neutrophils, B cells, immature and mature dendritic cells, regulatory T cells, natural killer T cells, and increased ratios of CD4/CD8 or Th2/Th1 T cells have been described as participants of this process [363,364,365].
Antibodies that inhibit immunological checkpoint molecules were conceived based on the hypothesis that blocking immune inhibitory signals could restore effector activity of T cells or boost T cell priming. Accordingly, checkpoint inhibitors interrupt the inhibition signaling of checkpoint molecules, promoting antitumor effector responses. With preclinical and clinical advances, antibodies such as ipilimumab (anti-CTLA-4 antibody), nivolumab, pembrolizumab (anti-PD-1 antibodies), durvalumab and avelumab (anti-PD-L1 antibodies) have been approved by the United States’ Food and Drug Administration (FDA). Several other antibodies targeting other immunological checkpoint molecules are under development and have shown promising results against several types of cancer [366]. In the last decade, immunotherapy revolutionized cancer therapy and is currently a centerpiece of adjuvant and neoadjuvant therapies, by itself or combined to chemotherapy and/or radiotherapy [367, 368].
Immunotherapy response-determinant host factors: you change, immunotherapy changes
In addition to understanding the TME, recent studies have also demonstrated the importance of patient-associated factors that can impact the response to immunotherapy, such as body composition (fat mass, fat-free mass and muscle quality), sex, ethnicity, age, habits (e.g., smoking, alcohol use, physical activity), past illnesses (immune history) and gut microbiota [23, 24, 369,370,371,372,373]. Obesity, currently considered a pandemic, is characterized by changes in the physiology of the entire body. Compromised body homeostasis due to obesity is associated with unregulated immune responses and chronic systemic inflammation, involving inflammatory factors such as leptin [374,375,376,377]. Paradoxically, these changes can be associated either with the appearance and progress of malignancies or as a protective phenomenon in certain types of tumors. These changes can be understood as the appearance of tumor cell phenotypes that can be more or less favored in disturbed systems, such as in patients with obesity [375, 378, 379]. Recent studies have mainly demonstrated the correlation between obesity and the response rate to immunotherapies of immune checkpoint inhibitors [199, 377, 380].
Obesity as a factor in immunotherapy success: hitting the checkpoint
Obesity impacts several features of T cell responses, aiding the immune escape of tumors through many mechanisms. It has been shown that in obesity, there is a decrease in the proliferative capacity of T cells, production of interferon gamma (IFNγ) and TNF-α, and increased expression of PD-1, which may be associated with leptin signaling [377]. Signaling promoted by leptins leads to increased pro-inflammatory T-helper 1 (Th1) immune responses, and its relationship with increased exhaustion and T cell dysfunction in obese patients is probably due to a phospho-signal mediated by the transducer and activation of transcription 3 (STAT3) pathway [268, 376, 381, 382]. Wang et al. demonstrated that, in obese animals, changes in PD-1 expression may contribute to a higher response rate to immune checkpoint blockade (ICB) therapy with PD-1/PD-L1 inhibitors, leading to increased infiltration of CD8+ T cells in subcutaneous tumors, reduction in the number of lung metastases and increased overall and progression-free survival. In human colorectal cancers, a low response to immunotherapy with anti-PD-1/PD-L1 is generally observed, and it has been shown that there is a smaller number of T cells in the tumor of obese compared to non-obese patients. In another cohort of patients with melanoma, a tumor with favorable of response to ICBs, there was a significant increase in the expression of PD-1 in tumors of obese patients. In obese animals, increased tumor progression has also been demonstrated, probably due to the induction of an immunosuppressive state and effects mediated by metabolites and hormones, such as leptin [377]. The chronic inflammation process, frequently observed in obese patients, can also induce immunosuppression, a protection mechanism against possible autoreactive responses of the immune system.
Immunotherapy and the obesity paradox
Obesity paradox in immunotherapy: a place to call home?
When trying to establish hypothesis and cause-consequence effects of the obesity paradox, an obstacle is the fact that obesity itself is multi-faceted and induces macro, systemic physiological alterations, making it difficult to determine beginning and endpoints for the observed associations with cancer development and treatment. By focusing observations of the obesity paradox on more micro, specific pathways, mechanisms can be more thoroughly analyzed.
It has been well established that obesity can trigger severe modulation of the immune landscape, resulting in metabolic and immunological dysfunction. In this way, adipose tissue from individuals with obesity can display immunological and metabolic modifications that can deeply impact cancer developing, as well as antitumor immunotherapies, since the chronic low inflammatory state associated with obesity has varied effects on anti-cancer immunity and immunotherapy effectiveness. Indeed, adipose tissues can be highly plastic and strongly impact inflammatory response and cancer progression by several different mechanisms [18]. Obesity can accelerate thymic aging, compromising T lymphocytes formation and proliferation, and consequently impairing progenitor pool and generation of the restricting the T cell repertoire [383]. Moreover, people with obesity can often display T lymphocytes with an exhausted phenotype in phenomenon that can be orchestrated by adipocytes and macrophages as a result of chronic inflammation due to prolonged stimulation of toll-like receptors by circulating free fatty acids, adipocyte cell death, activated stress responses, and hypoxia [384].
At first, obesity was thought to increase toxicity and impair immune efficacy, which makes sense with observations of T-cell inflammaging-like processes in a many animal species and, particularly, humans [377]. It has been showed that obesity induces T cell dysfunction and an upregulation of PD-1 on T lymphocytes, in a partially leptin-dependent manner. However, the polarization of T cells towards an exhaustive phenotype is correlated with improved response rates to anti-PD-1 therapy in the setting of obesity. Indeed, recent studies comparing obese to lean patients undergoing immunotherapeutic treatment show improved response to immunotherapy, together with longer progression-free and overall survival [385]. These obesity-favored effects were specially observed in checkpoint blockade targeting PD-1/PD-L1 of patients with multiple cancer types [199, 377] (Fig. 3). Therefore, obesity, which is associated with T cell dysfunction and worsen cancer prognostic, also paradoxically induces a better response to anti-PD-1/PD-L1 immunotherapy [386,387,388].

Implications of obese adipose tissue upon the immune system and immune checkpoint proteins. The dysregulation of the secretion of adipokines, free fatty acids and pro-inflammatory cytokines by the adipose tissue from individuals with obesity has a number of implications upon the immune system. These factors regulate both the proliferation of T cells, as well as their exhaustion, via modulation of PD-1 and PD-L1 expression, also affecting the antitumor immune response against cancer and immune checkpoint inhibitor immunotherapy. The polarization of macrophages from a M2 to a M1 profile induces changes in inflammatory cytokines production, often resulting in a ‘cytokine storm’, an event of uncontrolled inflammation which results in toxicity and failure of different organs
In the context of this obesity paradox, different mice models resulted from genetic ablation or diets can be used to understand better the role of obesity in cancer progression and anti-tumor immunotherapy during preclinical studies. Diet-induced obesity animal models can be frequently established by feeding animals with different diets containing for instance a high proportion of sugar (high-sugar diet) or fat (high-fat diet), or a combination of high-fat and high-carbohydrates in a diet called, cafeteria diet [389,390,391]. Amongst genetic animal models, mice with deficient leptin signaling are the most regularly used [262]. In this model, obesity can be induced by a mutation in the leptin receptor gene (db/db mouse) or by the lack of leptin (the ob/ob mouse), both causing the mice to overfeed.
Murphy and colleagues identified leptin as a potential therapeutic target for neutralization to enhance immunotherapy efficacy in obese cancer patients [392]. By using a leptin-deficient (ob/ob) obese BALB/c mice fed a high-fat diet model these authors demonstrated that systemic anti–CTLA-4 mAb and intratumoral delivery of a TRAIL-encoding adenovirus plus CpG immunnotherapies were effective in lean mice, but not in diet-induced obese BALB/c mice [392]. These data highlighted the potential of targeting leptin to boost cancer immunotherapy when in individuals with obesity.
Genetic mice models which allow a rapid development of highly metastatic tumors can be also used to study the obesity paradox in cancer development. Using a MMTV-PyMT mice as breast cancer model, Cranford and colleagues showed that high fat diet-induced obesity lead to an increased hormone production via aromatase expression and inflammation, enhancing breast cancer tumorigenesis [393]. Moreover, the MMTV-PyMT mice model has been used to test CTLA-4 or PD-1 checkpoint inhibition effects [394]. Additionally, Gibson and colleagues also investigated the effect of obesity on MDSC-mediated immunotherapy resistance in a breast cancer mouse tumor model, showing that obesity could trigger the accumulation of FasL+ granulocytic MDSCs, inducing apoptosis of tumor-infiltrating CD8 T lymphocytes and immunotherapy resistance in this cancer [395].
There is complex balance among diet, gut microbiota diversity, obesity, and the regulation of immune and inflammatory responses which can directly impact cancer development and antitumor immunotherapy. Obesity, gut microbiota and inflammatory signaling pathways are deeply connected. It has been showed that mice fed a high-fat diet alters gut microbiota [396] and mice lacking inflammasome’s caspases 1/11, but not NLRP3, were more susceptible to high fat diet-induced weight gain and presented higher abundance of phyla related to inflammation and gut dysbiosis, compared to wild type mice [397]. In another study, gut dysbiosis and high-fat diet present in a mice with predisposition to develop intestinal cancer (KrasG12Dint) accelerated the development of intestinal tumors [398]. In fact, gut microbiota function as strong regulator of antitumor immune response. Administration of antibiotics and fecal microbiota transplantation might impact the efficacy and toxicity of immunotherapy via gut microbiota [399,400,401].
Obesity paradox in melanoma immunotherapy: right under my skin
Currently, melanoma is highly targeted by immunotherapies, with seven different FDA-approved options for treating this type of cancer. These include oncolytic virus therapy and immune modulators, with marked observations using immune checkpoint inhibitors, such as ipilimumab, pembrolizumab, and nivolumab (targeting CTLA-4 and PD-1), or a combination of these [402, 403]. Notably, high BMI immunotherapy-treated melanoma patients show favorable outcomes, where several clinical studies report favorable associations between high BMI and immune checkpoint therapy in the context of melanoma, with or without accompanying chemotherapy [385, 404]. In these situations, overweight melanoma patients, when compared to lean patients, show better progression-free and overall survival. Of note, there is no difference in observations using PD-1 or PD-L1 immunotherapy [199, 377, 387, 402, 405].
In animal models of obese melanoma-bearing mice, anti-PD-1 immunotherapy enhances response to cancer by increasing number and function of tumor-associated CD8+ T cells, alongside a decrease in PD-1 expression by T cells, implying that the blockade of PD-1 is able to overcome obesity-driven T-cell exhaustion [377]. Leptin has also been associated with PD-1 expression and T-cell exhaustion. Leptin-induced TME immunosuppression is mediated by increased PD-1 expression and upregulation of activated STAT3, a mediator of leptin signaling which interacts with the PD-1 gene promoter [406]. On the other hand, in mice models of obesity, there was no improvement of anti-CTLA-4 treatment response until leptin was neutralized using soluble mouse leptin receptors, which lead to an increase in co-stimulatory CD86 expression [392]. This contradictory effect of leptin depending on the target of immunotherapy reveals the complexity of leptin signaling in innate and adaptive immune responses [407]. Obesity-induced alterations in gut microbiota have also been linked with the efficacy of ICB [372, 408].
Another important factor in determining favorable response to anti-melanoma immunotherapy is sex-related differences in hormone production, mainly estrogen, already implicated in the regulation of innate and adaptive immunity [409]. Indeed, female melanoma patients outperform males when comparing their response to immunotherapy [410, 411]. Even though melanoma does not express classical estrogen receptors, it does express G-protein-coupled estrogen receptors (GPERs), which, when activated, decrease PD-L1 expression and increase tumor susceptibility to T cells [412]. Studies regarding anti-PD-1 immunotherapy in mice showed synergy between ICB and GPERs activation, resulting in tumor regression, extended survival, and improved immune memory [412, 413].
Obesity paradox in NSCLC and renal cell carcinoma immunotherapy: it fits
A common trend of the obesity paradox in cancer is being true in a few specific cases but failing to address the benefits of obesity in the majority of cancer types. Since data availability of the correlation between BMI and immunotherapy efficacy is still very much scarce, no definitive conclusion about the extent of obesity paradox in immunotherapy can be given. Nonetheless, recent literature does reflect its existence and benefits in non-melanoma cancer, such as NSCLC, and renal cell carcinoma.
Throughout this review, evidence already pointed to the benefits of obesity in NSCLC when analyzing other parameters than response to immunotherapy. Although descriptions of the obesity paradox in lung cancer are not so mechanistically profound, epidemiological evidence is plenty. At least five different studies report a positive association between obesity (especially subcutaneous fat mass) and improved outcomes in patients receiving immune checkpoint inhibitors [414]. When compared to lean individuals, obese patients treated with pembrolizumab, nivolumab, or atezolizumab showed better and improved response to treatment, prolonged progression-free survival, and increased overall survival. These results are also true for advanced stages of lung cancer [415,416,417,418]. Together, they point to the existence of the obesity paradox in lung cancer immunotherapy, similarly to melanoma. Of note, Magri et al. used computerized tomography to analyze their data, which confirmed results had with BMI alone [419]. Curiously, no correlation between BMI and improved outcomes were observed for patients treated with chemotherapy, suggesting that, in lung cancer, at least, the clinical benefits of the obesity paradox are unique to immunotherapy [415].
Renal cell carcinoma (RCC) is another cancer subset reported to fit in the obesity paradox of cancer immunotherapy [420]. Three different studies comparing obese and lean patients treated with nivolumab or ipilimumab showed BMI had positive association with overall progression-free and overall survival, even when no correlation between BMI overall response rate, and disease control rate was found [421,422,423]. Conflicting reports, however, do exist. A study by Boi et al. associated obesity to worst overall and progression-free survival in RCC patients receiving standard anti-PD-1 immunotherapy [424].
Obesity paradox in breast cancer immunotherapy: the host’s contribution
The efficiency of anti-PD-1 immunotherapy for breast cancer (particularly TNBC) is sparse [425]. In fact, one of the few reports in which obesity is associated with better progression-free and overall survival of breast cancer patients reports the use of different immunotherapeutics, in this case, bevacizumab, an inhibitor of VEGF-A [260].
An obstacle for the success of anti-PD-1 immunotherapies in breast cancer is the recently described production of PD-L1 by the host’s adipose tissue [425]. This marked feature of breast cancer patients adds to the complexity of the obesity paradox in cancer immunotherapy. Breast cancer TME is an adipocyte-rich environment, therefore, secretion from adipocytes is particularly important. Proposed mechanisms for the interference of adipose PD-L1 with anti-PD-1/PD-L1 immunotherapy include binding of adipocyte PD-L1 to anti-PD-L1 antibodies, and interaction of adipocyte PD-L1 with T-cell PD-1 directly. Accordingly, knockout models for adipocyte PD-L1 alter T cell activation and tumor killing [426, 427]. Yet, the FDA recently approved the use of anti-PD-L1 atezolizumab for TNBC, and anti-CTLA-4 immunotherapies are currently under clinical trials [428].
Immune-related adverse events: immunotherapy success is not for free
Overall, immunotherapy is a successful, promising, and more personalized treatment for cancer patients. Regardless of its efficacy, immunotherapy, by its own purpose and immunosuppressive action mechanisms, can induce adverse events, denominated immune-related adverse events (irAEs). The immune checkpoints upon which immunotherapy exerts its effects are spread over many different immune cells (Treg cells, MDSCs, γδ T cells, TAMs, etc.), and their blockage alters the immune homeostasis, possibly leading to a series of autoimmune incidents [429]. Unbalance of T effector/T regulatory cells ratios and overactivation of T lymphocytes (including cytotoxic T cells) may provoke killing of normal, non-transformed cells, induce the release of neoantigens, tumor antigens and auto-antigens, and result in the release of large amounts of pro-inflammatory cytokines (IFN-γ, IL-17 and IL-6, from Th1 and Th17 T lymphocytes), culminating in a lethal cytokine storm [430,431,432,433].
Obesity-associated inflammation can also impact the immune response and consequently affect the efficacy and toxicity of immunotherapy. Apart from the beneficial aspects of obesity in cancer immunotherapy, the persistence of inflammation in obese patients has been linked to the development of irAEs [30]. Indeed, some studies associated higher BMI with increased risks of irAEs after immunotherapy [414, 434, 435]. In addition, limited evidence is available on the efficacy of immunotherapy upon obese and lean patients, which draws attention for considering BMI when designing randomized clinical trials for newer therapies [321].
Conclusion
The established link between obesity and several types of cancers raise important questions regarding conversion of this knowledge into actual and effective protective measures against cancer. Undoubtedly, maintenance of a healthy weight is an evident stage to potentially reducing risk of obesity-related cancers. However, even though elevated BMI is associated with increased cancer incidence for several cancer types, a number of studies have emerged evidence that cancer patients with obesity can present an improved survival rate upon immunotherapy against cancer. Although, this therapeutic advantage of a higher BMI is observed in few cancers during anti-tumor immunotherapy, millions of patients with obesity and cancer could benefit from this increasing evidence about the underlying physiological and biological mechanisms of obesity paradox’s effects upon cancer treatment and survival.
Availability of data and materials
Not applicable.
Abbreviations
- (AMP):
-
Adenosine monophosphate
- AGE:
-
Advanced glycation end product
- AIM2:
-
Absent in melanoma 2
- AMPK :
-
Activate 5′-adenosine monophosphate activated protein kinase
- APC:
-
Antigen presenting cells
- AT:
-
Adipose tissue
- ATM:
-
Tissue microenvironment
- ATP:
-
Adenosine triphosphate
- BAT:
-
Brown adipose tissues
- BMI:
-
Body Mass Index
- CAA:
-
Cancer-associated adipocytes
- CD:
-
Cluster of differentiation
- CLS:
-
Crown-like structures
- COX2:
-
Cyclooxygenase 2
- CTLA-4:
-
Cytotoxic T lymphocyte associated protein 4
- CVD:
-
Cardiovascular disease
- DAMP:
-
Damage-associated molecular patterns
- ER:
-
Endoplasmic reticulum
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal-regulated kinases
- ETC:
-
Electron transport chain
- FDA:
-
Food and Drug Administration
- FFA:
-
Free fatty acids
- GPERs:
-
G-protein-coupled estrogen receptors
- GSDMD:
-
Gasdermin D
- HFD:
-
High-fat diet
- HIF-1a:
-
Hypoxia-inducible factor 1-alpha
- HMGB1:
-
High Mobility Group Box-1
- ICB:
-
Immune checkpoint blockade
- IFNγ:
-
Interferon-gamma
- IL:
-
Interleukin
- ILC:
-
Lymphoid cells
- IR:
-
Insulin resistance
- irAE:
-
Immune-related adverse event
- JAK:
-
Janus kinase
- JNK:
-
C-Jun N-terminal kinase
- LAG3:
-
Lymphocyte-activation gene 3
- LPS:
-
Lipopolysaccharide
- LTB4 :
-
Leukotriene B4
- LTB4 :
-
Leukotriene B4
- MCP1:
-
Monocyte chemoattractant protein-1
- MDSC:
-
Myeloid-derived suppressor cells
- MHNO:
-
Metabolically healthy non-obese
- MHO:
-
Metabolically healthy obese
- MMTV-PyMT:
-
Mouse mammary tumor virus-polyoma middle tumor-antigen
- MS:
-
Metabolic syndrome
- mTOR:
-
(Mammalian target of rapamycin)
- MUNO:
-
Metabolically unhealthy non-obese individual
- MUO:
-
Unhealthy obese individual
- NAFLD:
-
Non-alcoholic fatty liver diseases
- NF-κB:
-
Nuclear factor kappa-light chain-enhancer of activated B cells
- NK:
-
Natural killer
- NLR:
-
NOD-like receptors
- NLRP3:
-
NOD-like receptor protein-3
- NOD:
-
Nucleotide-binding oligomerization domain
- NSCLC:
-
Non-small cell lung cancer
- OCT4:
-
Octamer-binding transcription factor 4
- PAI-1:
-
Plasminogen activator inhibitor-1
- PD-1:
-
Programmed death receptor 1
- PDL-1:
-
Programmed death receptor ligand 1
- PI3K:
-
Phosphoinositide 3-kinase
- RAGE:
-
Receptor for advanced glycation end product
- RCC:
-
Renal cell carcinoma
- ROS:
-
Reactive oxygen species
- SAT:
-
Subcutaneous adipose tissue
- SOX2:
-
Sex determining region Y-box 2
- STAT:
-
Signal transducer and activator of transcription proteins
- T2D:
-
Type 2 diabetes
- TAM:
-
Tumor associated macrophages
- TCR:
-
T cell antigen receptor
- Th:
-
Helper T lymphocytes
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containg-3
- TLR:
-
Toll-like receptors
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- TNF-α:
-
Tumor necrosis factor-alpha
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- Treg:
-
Regulatory T lymphocytes
- VAT:
-
Visceral adipose tissue
- VAT:
-
Visceral adipose tissue
- VEGF:
-
Vascular endothelial growth factor
- VISTA:
-
V-domain Ig suppressor of T cell activation
- WAT:
-
White adipose tissue
References
Swinburn BA, Kraak VI, Allender S, Atkins VJ, Baker PI, Bogard JR, et al. The global syndemic of obesity, undernutrition, and climate change: the Lancet commission report. Lancet. 2019;393:791–846.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Li J, Ma J, Wang KS, Mi C, Wang Z, Piao LX, et al. Baicalein inhibits TNF-α-induced NFkB activation and expression of NFkB-regulated target gene products. Oncol Rep. 2016;36:2771–6.
National Cancer Institute, NIH D. Cancer trends progress report. National Cancer Institute. 2020.
Tremmel M, Gerdtham U-G, Nilsson P, Saha S. Economic burden of obesity: a systematic literature review. Int J Environ Res Public Health. 2017;14:435.
Temple NJ, Conklin A. Prevalence of overweight and obesity in Western countries: discrepancies in published estimates. Eur J Epidemiol. 2019;34:711–3.
Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–91.
Brawley OW. Trends in prostate cancer in the United States. JNCI Monogr. 2012;2012:152–6.
Thun MJ, DeLancey JO, Center MM, Jemal A, Ward EM. The global burden of cancer: priorities for prevention. Carcinogenesis. 2010;31:100–10.
World Health Organization. WHO report on cancer: setting priorities, investing wisely and providing care for all. Geneva: World Health Organization; 2020.
Yabroff KR, Lund J, Kepka D, Mariotto A. Economic burden of cancer in the United States: estimates, projections, and future research. Cancer Epidemiol Biomark Prev. 2011;20:2006–14.
Hong Y-R, Huo J, Desai R, Cardel M, Deshmukh AA. Excess costs and economic burden of obesity-related cancers in the United States. Value Health. 2019;22:1378–86.
Lee JE, Nam CM, Lee SG, Park S, Kim TH, Park E. The economic burden of cancer attributable to obesity in Korea: a population-based cohort study. Eur J Cancer Care (Engl). 2019;28:1–10.
Kolb R, Sutterwala FS, Zhang W. Obesity and cancer: inflammation bridges the two. Curr Opin Pharmacol. 2016;29:77–89.
De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obes. 2013;2013:291546.
Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, Abed Y. Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci. 2017;4:851–63.
Ramos-Nino ME. The role of chronic inflammation in obesity-associated cancers. ISRN Oncol. 2013;2013:1–25.
Corrêa, Heyn, Magalhaes. The impact of the adipose organ plasticity on inflammation and cancer progression. Cells 2019;8:662.
Zhang T, Kolonin MG. Role of adipose cells in tumor microenvironment. In: Gefen A, Benayahu D, editors. The mechanobiology of obesity and related diseases. Studies in mechanobiology, tissue engineering and biomaterials, vol 16. Cham: Springer. 2013. https://doi.org/10.1007/8415_2013_171.
Hefetz-Sela S, Scherer PE. Adipocytes: impact on tumor growth and potential sites for therapeutic intervention. Pharmacol Ther. 2013;138:197–210.
Kothari C, Diorio C, Durocher F. the importance of breast adipose tissue in breast cancer. Int J Mol Sci. 2020;21:5760.
Lu J, Zhao J, Meng H, Zhang X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front Immunol. 2019;10:1–12.
Trestini I, Carbognin L, Bonaiuto C, Tortora G, Bria E. The obesity paradox in cancer: clinical insights and perspectives. Eat Weight Disord. 2018;23:185–93.
Lennon H, Sperrin M, Badrick E, Renehan AG. The obesity paradox in cancer: a review. Curr Oncol Rep. 2016;18:56.
Gonzalez MC, Pastore CA, Orlandi SP, Heymsfield SB. Obesity paradox in cancer: new insights provided by body composition. Am J Clin Nutr. 2014;99:999–1005.
Sinicrope FA, Foster NR, Yothers G, Benson A, Seitz JF, Labianca R, et al. Body mass index at diagnosis and survival among colon cancer patients enrolled in clinical trials of adjuvant chemotherapy. Cancer. 2013;119:1528–36.
Demark-Wahnefried W, Platz EA, Ligibel JA, Blair CK, Courneya KS, Meyerhardt JA, et al. the role of obesity in cancer survival and recurrence. Cancer Epidemiol Biomark Prev. 2012;21:1244–59.
Zhang X, Liu Y, Shao H, Zheng X. Obesity paradox in lung cancer prognosis: evolving biological insights and clinical implications. J Thorac Oncol. 2017;12:1478–88.
Suzuki Y, Okamoto T, Fujishita T, Katsura M, Akamine T, Takamori S, et al. Clinical implications of sarcopenia in patients undergoing complete resection for early non-small cell lung cancer. Lung Cancer. 2016;101:92–7.
Naik A, Monjazeb AM, Decock J. The obesity paradox in cancer, tumor immunology, and immunotherapy: potential therapeutic implications in triple negative breast cancer. Front Immunol. 2019;10:1–12.
Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. 2019;15:139–54.
Rojas A, Araya P, Gonzalez I, Morales E. Tumor microenvironments in organs. Birbrair A, editor. Adv Exp Med Biol. 2020;1226:23–35.
Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–7.
McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48.
Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542:177–85. https://doi.org/10.1038/nature21363.
Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. 2017;127:43–54.
Rochlani Y, Pothineni NV, Kovelamudi S, Mehta JL. Metabolic syndrome: pathophysiology, management, and modulation by natural compounds. Ther Adv Cardiovasc Dis. 2017;11:215–25.
Kanwar P, Kowdley KV. The metabolic syndrome and its influence on nonalcoholic steatohepatitis. Clin Liver Dis. 2016;20:225–43.
Gierach M, Gierach J, Ewertowska M, Arndt A, Junik R. Correlation between body mass index and waist circumference in patients with metabolic syndrome. ISRN Endocrinol. 2014;2014:1–6.
Lukács A, Horváth E, Máté Z, Szabó A, Virág K, Papp M, et al. Abdominal obesity increases metabolic risk factors in non-obese adults: a Hungarian cross-sectional study. BMC Public Health. 2019;19:1–8.
Cartier A, Côté M, Bergeron J, Alméras N, Tremblay A, Lemieux I, et al. Plasma soluble tumour necrosis factor-α receptor 2 is elevated in obesity: specific contribution of visceral adiposity. Clin Endocrinol (Oxf). 2010;72:349–57.
Chandra A, Neeland IJ, Berry JD, Ayers CR, Rohatgi A, Das SR, et al. The relationship of body mass and fat distribution with incident hypertension: observations from the Dallas Heart Study. J Am Coll Cardiol. 2014;64:997–1002.
Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48.
Engin A. The definition and prevalence of obesity and metabolic syndrome. Adv Exp Med Biol. 2017;960:1–17. https://doi.org/10.1007/978-3-319-48382-5_1.
Oda E. Historical perspectives of the metabolic syndrome. Clin Dermatol. 2018;36:3–8.
Roberts CK, Hevener AL, Barnard RJ. Metabolic syndrome and insulin resistance: underlying causes and modification by exercise training. Compr Physiol. 2013;3:1–58.
Prasun P. Mitochondrial dysfunction in metabolic syndrome. Biochim Biophys Acta. 2020;1866:165838.
Mohan S, Preetha Rani MR, Brown L, Ayyappan P, Raghu KG. Endoplasmic reticulum stress: a master regulator of metabolic syndrome. Eur J Pharmacol. 2019;860:172553.
Cunarro J, Casado S, Lugilde J, Tovar S. Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease. Front Endocrinol (Lausanne). 2018;9:1–10.
Walsh CT, Tu BP, Tang Y. Eight kinetically stable but thermodynamically activated molecules that power cell metabolism. Chem Rev. 2018;118:1460–94.
Brookes PS. Mitochondrial H + leak and ROS generation: an odd couple. Free Radic Biol Med. 2005;38:12–23.
Bergman O, Ben-Shachar D. Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: possible interactions with cellular processes. Can J Psychiatry. 2016;61:457–69.
Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med. 1999;26:463–71.
Betteridge DJ. What is oxidative stress? Metabolism. 2000;49:3–8.
Quirós PM, Langer T, López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16:345–59.
Li X. Endoplasmic reticulum stress regulates inflammation in adipocyte of obese rats via toll-like receptors 4 signaling. Iran J Basic Med Sci. 2018;21:502–7.
Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73:79–94.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29.
Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–49.
Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci. 2019;6:1–12.
O’Rourke RW. Inflammation in obesity-related diseases. Surgery. 2009;145:255–9.
Thoudam T, Jeon JH, Ha CM, Lee IK. Role of mitochondria-associated endoplasmic reticulum membrane in inflammation-mediated metabolic diseases. Mediat Inflamm. 2016;2016:10–3.
Masschelin PM, Cox AR, Chernis N, Hartig SM. The impact of oxidative stress on adipose tissue energy balance. Front Physiol. 2020;10:1–8.
Okuno Y, Fukuhara A, Hashimoto E, Kobayashi H, Kobayashi S, Otsuki M, et al. Oxidative stress inhibits healthy adipose expansion through suppression of SREBF1-mediated lipogenic pathway. Diabetes. 2018;67:1113–27.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91.
Christiansen T, Richelsen B, Bruun JM. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes. 2005;29:146–50.
Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, et al. Interferon-γ, a Th1 cytokine, regulates fat inflammation. Circ Res. 2008;103:467–76.
Jiang E, Perrard XD, Yang D, Khan IM, Perrard JL, Smith CW, et al. Essential role of CD11a in CD8 + T-cell accumulation and activation in adipose tissue. Arterioscler Thromb Vasc Biol. 2014;34:34–43.
Li P, Young Oh D, Bandyopadhyay G, Lagakos WS, Talukdar S, et al. LTB4 causes macrophage–mediated inflammation and directly induces insulin resistance in obesity. Nat Med. 2015;21:239–47.
Hadad N, Burgazliev O, Elgazar-Carmon V, Solomonov Y, Wueest S, Item F, et al. Induction of cytosolic phospholipase a2á is required for adipose neutrophil infiltration and hepatic insulin resistance early in the course of high-fat feeding. Diabetes. 2013;62:3053–63.
Chen A, Mumick S, Zhang C, Lamb J, Dai H, Weingarth D, et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes Res. 2005;13:1311–20.
Keophiphath M, Rouault C, Divoux A, Clément K, Lacasa D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol. 2010;30:39–45.
Su X, Peng D. Adipokines as novel biomarkers of cardio-metabolic disorders. Clin Chim Acta. 2020;507:31–8.
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9.
Pereira S, Teixeira L, Aguilar E, Oliveira M, Savassi-Rocha A, Pelaez JN, et al. Modulation of adipose tissue inflammation by FOXP3+ Treg cells, IL-10, and TGF-β in metabolically healthy class III obese individuals. Nutrition. 2014;30:784–90.
Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in high fat diet fed mice via secreted elastase. Nat Med. 2012;18:1407–12.
Gurung P, Moussa K, Adams-Huet B, Devaraj S, Jialal I. Increased mast cell abundance in adipose tissue of metabolic syndrome: relevance to the proinflammatory state and increased adipose tissue fibrosis. Am J Physiol Endocrinol Metab. 2019;316:E504–9.
Lee B-C, Kim M, Pae M, Yamamoto Y, Eberlé D, Shimada T, et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab. 2016;23:685–98.
O’Sullivan TE, Rapp M, Fan X, Weizman O-E, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. 2016;45:428–41.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20.
Yang H, Youm Y, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. 2010;185:1836–45.
Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–7.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808.
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84.
Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. 2008;49:1562–8. https://doi.org/10.1194/jlr.M800019-JLR200.
McDonnell ME, Ganley-Leal LM, Mehta A, Bigornia SJ, Mott M, Rehman Q, et al. B lymphocytes in human subcutaneous adipose crown-like structures. Obesity. 2012;20:1372–8.
Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, et al. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem. 2010;285:3428–38.
Zhang J, Zhang L, Zhang S, Yu Q, Xiong F, Huang K, et al. HMGB1, an innate alarmin, plays a critical role in chronic inflammation of adipose tissue in obesity. Mol Cell Endocrinol. 2017;454:103–11.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–98.
Liang F, Zhang F, Zhang L, Wei W. The advances in pyroptosis initiated by inflammasome in inflammatory and immune diseases. Inflamm Res. 2020;69:159–66.
Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87. https://doi.org/10.1038/nm.3893.
Wagner M. A dangerous duo in adipose tissue: high-mobility group box 1 protein and macrophages. Yale J Biol Med. 2014;87:127–33.
Rockenfeller P, Ring J, Muschett V, Beranek A, Buettner S, Carmona-Gutierrez D, et al. Fatty acids trigger mitochondrion-dependent necrosis. Cell Cycle. 2010;9:2908–14.
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The p2x7 receptor in infection and inflammation. Immunity. 2017;47:15–31.
Lee YS, Kim J, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell. 2014;157:1339–52.
Fujisaka S, Usui I, Ikutani M, Aminuddin A, Takikawa A, Tsuneyama K, et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1α-independent manner in obese mice. Diabetologia. 2013;56:1403–12.
Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the international diabetes federation task force on epidemiology and prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation International. Circulation. 2009;120:1640–5.
Piperi C, Adamopoulos C, Dalagiorgou G, Diamanti-Kandarakis E, Papavassiliou AG. Crosstalk between advanced glycation and endoplasmic reticulum stress: emerging therapeutic targeting for metabolic diseases. J Clin Endocrinol Metab. 2012;97:2231–42.
Kang R, Chen R, Xie M, Cao L, Lotze MT, Tang D, et al. The receptor for advanced glycation endproducts (RAGE) activates the AIM2 inflammasome in acute pancreatitis. J Immunol. 2017;196:4331–7.
Mori K, Suzuki T, Igarashi T, Inoue K, Asahara T, Nomoto K, et al. Persistent hyperglycemia modulates gut immune function and microbiota in rats. J Intensive Care. 2015;3:1–12.
Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–83.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MTH, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15.
Nguyen MTA, Favelyukis S, Nguyen A-K, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–92.
Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020;20:40–54.
Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. 2012;142:1100-1101.e2.
Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. Gut microbiota and metabolic syndrome. World J Gastroenterol. 2014;20:16079–94.
Lteif AA, Han K, Mather KJ. Obesity, insulin resistance, and the metabolic syndrome: determinants of endothelial dysfunction in whites and blacks. Circulation. 2005;112:32–8.
Himsworth HP. Diabetes mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. 1936. Int J Epidemiol. 2013;42:1594–8.
Faloia E, Michetti G, De Robertis M, Luconi MP, Furlani G, Boscaro M. Inflammation as a link between obesity and metabolic syndrome. J Nutr Metab. 2012;2012:7.
Vona R, Gambardella L, Cittadini C, Straface E, Pietraforte D. Biomarkers of oxidative stress in metabolic syndrome and associated diseases. Oxid Med Cell Longev. 2019. https://doi.org/10.1155/2019/8267234.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494.
Stolarczyk E, Vong CT, Perucha E, Jackson I, Cawthorne MA, Wargent ET, et al. Improved insulin sensitivity despite increased visceral adiposity in mice deficient for the immune cell transcription factor T-bet. Cell Metab. 2013;17:520–33.
Lukic L, Lalic NM, Rajkovic N, Jotic A, Lalic K, Milicic T, et al. Hypertension in obese type 2 diabetes patients is associated with increases in insulin resistance and IL-6 cytokine levels: potential targets for an efficient preventive intervention. Int J Environ Res Public Health. 2014;11:3586–98.
Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. 2015;16:376–85.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30.
Liu W, Zhou X, Li Y, Zhang S, Cai X, Zhang R, et al. Serum leptin, resistin, and adiponectin levels in obese and non-obese patients with newly diagnosed type 2 diabetes mellitus: a population-based study. Medicine (United States). 2020;99:1–7.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97.
Stienstra R, Joosten LAB, Koenen T, van Tits B, van Diepen JA, van den Berg SAA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605.
Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol. 2018;15:721–37.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72.
Samuel VT, Shulman GI. Integrating mechanisms for insulin resistance. Cell. 2012;148:852–71.
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77.
Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7:1016–36.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Weir HK, Thompson TD, Soman A, Møller B, Leadbetter S. The past, present, and future of cancer incidence in the United States: 1975 through 2020. Cancer. 2015;121:1827–37.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. https://doi.org/10.3322/caac.21590.
Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68:31–54.
Kyrgiou M, Kalliala I, Markozannes G, Gunter MJ, Paraskevaidis E, Gabra H, et al. Adiposity and cancer at major anatomical sites: umbrella review of the literature. BMJ. 2017;356:1–10.
Kolb R, Zhang W. Obesity and breast cancer: a case of inflamed adipose tissue. Cancers (Basel). 2020;12:1686.
Arnold M, Pandeya N, Byrnes G, Renehan AG, Stevens GA, Ezzati M, et al. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol. 2015;16:36–46.
Whiteman DC, Wilson LF. The fractions of cancer attributable to modifiable factors: a global review. Cancer Epidemiol. 2016;44:203–21.
Eheman C, Henley SJ, Ballard-Barbash R, Jacobs EJ, Schymura MJ, Noone A-M, et al. Annual report to the nation on the status of cancer, 1975–2008, featuring cancers associated with excess weight and lack of sufficient physical activity. Cancer. 2012;118:2338–66.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. https://doi.org/10.1002/ijc.29210.
de Rezende LFM, Arnold M, Rabacow FM, Levy RB, Claro RM, Giovannucci E, et al. The increasing burden of cancer attributable to high body mass index in Brazil. Cancer Epidemiol. 2018;54:63–70.
Park Y, Peterson LL, Colditz GA. The plausibility of obesity paradox in cancer—point. Cancer Res. 2018;78:1898–903.
Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J Clin Oncol. 2016;34:4270–6. https://doi.org/10.1200/JCO.2016.67.4283.
Renehan AG, Zwahlen M, Egger M. Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat Rev Cancer. 2015;15:484–98.
Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. Longo DL, editor. N Engl J Med. 2017;376:254–66.
Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45.
Lohmann AE, Goodwin PJ, Chlebowski RT, Pan K, Stambolic V, Dowling RJO. Association of obesity-related metabolic disruptions with cancer risk and outcome. J Clin Oncol. 2016;34:4249–55.
Li S, Wang Z, Huang J, Fan J, Du H, Liu L, et al. Systematic review of prognostic roles of body mass index for patients undergoing lung cancer surgery: does the ‘obesity paradox’ really exist? Eur J Cardio-Thorac Surg. 2016;51:ezw386.
Li L, Kalantar-Zadeh K. Obesity that makes kidney cancer more likely but helps fight it more strongly. J Natl Cancer Inst. 2013;105:1848–9.
Ito R, Narita S, Huang M, Nara T, Numakura K, Takayama K, et al. The impact of obesity and adiponectin signaling in patients with renal cell carcinoma: a potential mechanism for the “obesity paradox.” Lee JW, editor. PLoS ONE. 2017;12:e0171615.
Choi Y, Park B, Jeong BC, Seo SI, Jeon SS, Choi HY, et al. Body mass index and survival in patients with renal cell carcinoma: a clinical-based cohort and meta-analysis. Int J Cancer. 2013;132:625–34.
Barth RF, Maximilian Buja L, Cao L, Brodsky SV. An obesity paradox: increased body mass index is associated with decreased aortic atherosclerosis. Curr Hypertens Rep. 2017;19:55.
Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol. 2017;14:133–44.
Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–97.
Lainscak M, von Haehling S, Doehner W, Anker SD. The obesity paradox in chronic disease: facts and numbers. J Cachexia Sarcopenia Muscle. 2012;3:1–4.
Park J, Ahmadi S-F, Streja E, Molnar MZ, Flegal KM, Gillen D, et al. Obesity paradox in end-stage kidney disease patients. Prog Cardiovasc Dis. 2014;56:415–25.
Doehner W, Erdmann E, Cairns R, Clark AL, Dormandy JA, Ferrannini E, et al. Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: an analysis of the PROactive study population. Int J Cardiol. 2012;162:20–6.
Brooks NA, Kokorovic A, Xiao L, Matulay JT, Li R, Ranasinghe WKB, et al. The obesity paradox: defining the impact of body mass index and diabetes mellitus for patients with non-muscle-invasive bladder cancer treated with bacillus Calmette–Guérin. BJU Int. 2020. https://doi.org/10.1111/bju.15296.
Goyal A, Nimmakayala KR, Zonszein J. Is there a paradox in obesity? Cardiol Rev. 2014;22:163–70.
Preston SH, Stokes A. Obesity paradox. Epidemiology. 2014;25:454–61.
Ferreira I, Stehouwer CDA. Obesity paradox or inappropriate study designs? Time for life-course epidemiology. J Hypertens. 2012;30:2271–5.
Stefan N, Häring HU, Hu FB, Schulze MB. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013;1:152–62.
Tian S, Liu Y, Feng A, Lou K, Dong H. Metabolically healthy obesity and risk of cardiovascular disease, cancer, and all-cause and cause-specific mortality: a protocol for a systematic review and meta-analysis of prospective studies. BMJ Open. 2019;9:e032742.
Janssen I, Katzmarzyk PT, Ross R. Waist circumference and not body mass index explains obesity-related health risk. Am J Clin Nutr. 2004;79:379–84.
Spahillari A, Mukamal KJ, DeFilippi C, Kizer JR, Gottdiener JS, Djoussé L, et al. The association of lean and fat mass with all-cause mortality in older adults: the Cardiovascular Health Study. Nutr Metab Cardiovasc Dis. 2016;26:1039–47.
Park YM, White AJ, Nichols HB, O’Brien KM, Weinberg CR, Sandler DP. The association between metabolic health, obesity phenotype and the risk of breast cancer. Int J Cancer. 2017;140:2657–66.
Ezzati M, Stevens GA, Finucane MM, Danaei G. Global trends in body-mass index—authors’ reply. Lancet. 2011;377:1917–8.
Stefan N, Kantartzis K, Machann J, Schick F, Thamer C, Rittig K, et al. Identification and characterization of metabolically benign obesity in humans. Obstet Gynecol Surv. 2009;64:30–1.
Wildman RP. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering. Arch Intern Med. 2008;168:1617.
Lin H, Zhang L, Zheng R, Zheng Y. The prevalence, metabolic risk and effects of lifestyle intervention for metabolically healthy obesity. Medicine (Baltimore). 2017;96:e8838.
Rey-López JP, de Rezende LF, Pastor-Valero M, Tess BH. The prevalence of metabolically healthy obesity: a systematic review and critical evaluation of the definitions used. Obes Rev. 2014;15:781–90.
Hamer M, Stamatakis E. Metabolically healthy obesity and risk of all-cause and cardiovascular disease mortality. J Clin Endocrinol Metab. 2012;97:2482–8.
Ärnlöv J, Ingelsson E, Sundström J, Lind L. Impact of body mass index and the metabolic syndrome on the risk of cardiovascular disease and death in middle-aged men. Circulation. 2010;121:230–6.
Yang HK, Han K, Kwon H-S, Park Y-M, Cho J-H, Yoon K-H, et al. Obesity, metabolic health, and mortality in adults: a nationwide population-based study in Korea. Sci Rep. 2016;6:30329.
Sahakyan KR, Somers VK, Rodriguez-Escudero JP, Hodge DO, Carter RE, Sochor O, et al. Normal-weight central obesity: implications for total and cardiovascular mortality. Ann Intern Med. 2015;163:827.
Ortega FB, Lee D, Katzmarzyk PT, Ruiz JR, Sui X, Church TS, et al. The intriguing metabolically healthy but obese phenotype: cardiovascular prognosis and role of fitness. Eur Heart J. 2013;34:389–97.
Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339:172–7.
Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806.
Kim J-Y, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–37.
Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313–25.
Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. 2012;18:1539–49.
Herman MA, Peroni OD, Villoria J, Schön MR, Abumrad NA, Blüher M, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–8.
Lavie CJ, Ventura HO. The obesity paradox in heart failure. JACC Heart Fail. 2015;3:927–30.
Lu Y, Hajifathalian K, Ezzati M, Woodward M, Rimm EB, Danaei G, et al. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1·8 million participants. Lancet. 2014;383:970–83.
Hashimoto Y, Hamaguchi M, Obora A, Kojima T, Fukui M. Impact of metabolically healthy obesity on the risk of incident gastric cancer: a population-based cohort study. BMC Endocr Disord. 2020;20:11.
Kim TJ, Kim ER, Hong SN, Kim Y-H, Chang DK, Ji J, et al. Metabolic unhealthiness is an important predictor for the development of advanced colorectal neoplasia. Sci Rep. 2017;7:9011.
Kim JW, Ahn ST, Oh MM, Moon DG, Cheon J, Han K, et al. Increased incidence of bladder cancer with metabolically unhealthy status: analysis from the National Health Checkup database in Korea. Sci Rep. 2020;10:6476.
Korduner J, Bachus E, Jujic A, Magnusson M, Nilsson PM. Metabolically healthy obesity (MHO) in the Malmö diet cancer study—epidemiology and prospective risks. Obes Res Clin Pract. 2019;13:548–54.
Kim TJ, Lee H, Baek S-Y, Kim K, Min YW, Min B-H, et al. Metabolically healthy obesity and the risk of erosive esophagitis: a cohort study. Clin Transl Gastroenterol. 2019;10:e00077.
Lassen A, Hallas J, de Muckadell OBS. Esophagitis: incidence and risk of esophageal adenocarcinoma—a population-based cohort study. Am J Gastroenterol. 2006;101:1193–9.
Lin C, Chang Y, Cheng T, Lo K, Liu S, Yeh TL. The association between metabolically healthy obesity and risk of cancer: a systematic review and meta-analysis of prospective cohort studies. Obes Rev. 2020;21:1–10.
Schulze MB. Metabolic health in normal-weight and obese individuals. Diabetologia. 2019;62:558–66.
Blüher M. Metabolically healthy obesity. Endocr Rev. 2020;41:405–20.
Aoyagi T, Terracina KP, Raza A, Matsubara H, Takabe K. Cancer cachexia, mechanism and treatment. World J Gastrointest Oncol. 2015;7:17.
Anker MS, Holcomb R, Muscaritoli M, Haehling S, Haverkamp W, Jatoi A, et al. Orphan disease status of cancer cachexia in the USA and in the European Union: a systematic review. J Cachexia Sarcopenia Muscle. 2019;10:22–34.
Rahman SU, Sana MK, Tahir Z, Ali A, Shah PA. Paraneoplastic syndromes in cholangiocarcinoma. World J Hepatol. 2020;12:897–907.
Ni J, Zhang L. Cancer cachexia: definition, staging, and emerging treatments. Cancer Manag Res. 2020;12:5597–605.
LeBlanc TW, Nipp RD, Rushing CN, Samsa GP, Locke SC, Kamal AH, et al. Correlation between the international consensus definition of the cancer anorexia-cachexia syndrome (CACS) and patient-centered outcomes in advanced non-small cell lung cancer. J Pain Symptom Manag. 2015;49:680–9.
Lüchtenborg M, Jakobsen E, Krasnik M, Linklater KM, Mellemgaard A, Møller H. The effect of comorbidity on stage-specific survival in resected non-small cell lung cancer patients. Eur J Cancer. 2012;48:3386–95.
Dhanapal R, Saraswathi T, Rajkumar G. Cancer cachexia. J Oral Maxillofac Pathol. 2011;15:257.
Delano MJ, Moldawer LL. The origins of cachexia in acute and chronic inflammatory diseases*. Nutr Clin Pract. 2006;21:68–81.
Deans C, Wigmore SJ. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nutr Metab Care. 2005;8:265–9.
Laviano A, Koverech A, Mari A. Cachexia: clinical features when inflammation drives malnutrition. Proc Nutr Soc. 2015;74:348–54.
Iachina M, Jakobsen E, Møller H, Lüchtenborg M, Mellemgaard A, Krasnik M, et al. The effect of different comorbidities on survival of non-small cells lung cancer patients. Lung. 2015;193:291–7.
McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol. 2018;19:310–22.
Fontaine KR, Redden DT, Wang C, Westfall AO, Allison DB. Years of life lost due to obesity. J Am Med Assoc. 2003;289:187–93.
Batsis JA, Villareal DT. Sarcopenic obesity in older adults: aetiology, epidemiology and treatment strategies. Nat Rev Endocrinol. 2018;14:513–37.
Roh E, Choi KM. Health consequences of sarcopenic obesity: a narrative review. Front Endocrinol (Lausanne). 2020;11:332.
Martin L, Birdsell L, MacDonald N, Reiman T, Clandinin MT, McCargar LJ, et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of Body Mass Index. J Clin Oncol. 2013;31:1539–47.
Demark-Wahnefried W, Campbell KL, Hayes SC. Weight management and its role in breast cancer rehabilitation. Cancer. 2012;118:2277–87.
Nattenmüller J, Wochner R, Muley T, Steins M, Hummler S, Teucher B, et al. Prognostic impact of CT-quantified muscle and fat distribution before and after first-line-chemotherapy in lung cancer patients. Singh PK, editor. PLoS ONE. 2017;12:e0169136.
Prado CM, Lieffers JR, McCargar LJ, Reiman T, Sawyer MB, Martin L, et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population-based study. Lancet Oncol. 2008;9:629–35.
Argilés JM, Busquets S, Toledo M, López-Soriano FJ. The role of cytokines in cancer cachexia. Curr Opin Support Palliat Care. 2009;3:263–8.
MacDonald N, Easson AM, Mazurak VC, Dunn GP, Baracos VE. Understanding and managing cancer cachexia. J Am Coll Surg. 2003;197:143–61.
Camargo R, Riccardi D, Ribeiro H, Carnevali L, de Matos-Neto E, Enjiu L, et al. NF-κBp65 and expression of its pro-inflammatory target genes are upregulated in the subcutaneous adipose tissue of cachectic cancer patients. Nutrients. 2015;7:4465–79.
Batista ML, Olivan M, Alcantara PSM, Sandoval R, Peres SB, Neves RX, et al. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine. 2013;61:532–9.
Neves RX, Rosa-Neto JC, Yamashita AS, Matos-Neto EM, Riccardi DMR, Lira FS, et al. White adipose tissue cells and the progression of cachexia: inflammatory pathways. J Cachexia Sarcopenia Muscle. 2016;7:193–203.
Batista ML, Neves RX, Peres SB, Yamashita AS, Shida CS, Farmer SR, et al. Heterogeneous time-dependent response of adipose tissue during the development of cancer cachexia. J Endocrinol. 2012;215:363–73.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Reynolds CM, McGillicuddy FC, Harford KA, Finucane OM, Mills KHG, Roche HM. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol Nutr Food Res. 2012;56:1212–22.
Benetti E, Chiazza F, Patel NSA, Collino M. The nlrp3 inflammasome as a novel player of the intercellular crosstalk in metabolic disorders. Mediat Inflamm. 2013;2013:1–9.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–89.
Stienstra R, Van Diepen JA, Tack CJ, Zaki MH, Van De Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USA. 2011;108:15324–9.
Ebadi M, Mazurak V. Evidence and mechanisms of fat depletion in cancer. Nutrients. 2014;6:5280–97.
Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014;20:433–47.
Lipina C, Hundal HS. Lipid modulation of skeletal muscle mass and function. J Cachexia Sarcopenia Muscle. 2017;8:190–201.
Andrich DE, Ou Y, Melbouci L, Leduc-Gaudet J-P, Auclair N, Mercier J, et al. Altered lipid metabolism impairs skeletal muscle force in young rats submitted to a short-term high-fat diet. Front Physiol. 2018;9:1–12.
Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–8.
Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, et al. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100–4.
Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Prim. 2018;4:17105.
Zimmermann R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–6.
Rohm M, Schäfer M, Laurent V, Üstünel BE, Niopek K, Algire C, et al. An AMP-activated protein kinase–stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat Med. 2016;22:1120–30.
Yang R, Cheung MC, Pedroso FE, Byrne MM, Koniaris LG, Zimmers TA. Obesity and weight loss at presentation of lung cancer are associated with opposite effects on survival. J Surg Res. 2011;170:e75-83.
Bandera EV, Fay SH, Giovannucci E, Leitzmann MF, Marklew R, McTiernan A, et al. The use and interpretation of anthropometric measures in cancer epidemiology: a perspective from the world cancer research fund international continuous update project. Int J Cancer. 2016;139:2391–7.
Molica F, Morel S, Kwak B, Rohner-Jeanrenaud F, Steffens S. Adipokines at the crossroad between obesity and cardiovascular disease. Thromb Haemost. 2015;113:553–66.
Diedrich J, Gusky HC, Podgorski I. Adipose tissue dysfunction and its effects on tumor metabolism. Horm Mol Biol Clin Invest. 2015;21:17–41.
Nimri L, Peri I, Yehuda-Shnaidman E, Schwartz B. Adipocytes isolated from visceral and subcutaneous depots of donors differing in bmi crosstalk with colon cancer cells and modulate their invasive phenotype. Transl Oncol. 2019;12:1404–15.
Zha JM, Di WJ, Zhu T, Xie Y, Yu J, Liu J, et al. Comparison of gene transcription between subcutaneous and visceral adipose tissue in chinese adults. Endocr J. 2009;56:935–44.
Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2010;11:11–8.
Villaret A, Galitzky J, Decaunes P, Esteve D, Marques M-A, Sengenes C, et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59:2755–63.
Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, et al. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013;17:644–56.
Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–28.
Avgerinos KI, Spyrou N, Mantzoros CS, Dalamaga M. Obesity and cancer risk: emerging biological mechanisms and perspectives. Metabolism. 2019;92:121–35.
Cabia B, Andrade S, Carreira MC, Casanueva FF, Crujeiras AB. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes Rev. 2016;17:361–76.
Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev. 2012;33:547–94.
Saygin C, Reizes O, Berger NA. Adipocytes, adipocytokines, and cancer. In: Reizes O, Berger N, editors. Adipocytokines, energy balance, and cancer. Energy balance and cancer, vol 12. Cham: Springer. 2017. https://doi.org/10.1007/978-3-319-41677-9_1.
Spyrou N, Avgerinos KI, Mantzoros CS, Dalamaga M. Classic and novel adipocytokines at the intersection of obesity and cancer: diagnostic and therapeutic strategies. Curr Obes Rep. 2018;7:260–75.
Madak-Erdogan Z, Band S, Zhao YC, Smith BP, Kulkoyluoglu-Cotul E, Zuo Q, et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019;79:canres.2849.2018.
Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obes Rev. 2012;13:1083–95.
Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41–5.
Schwartz B, Yehuda-Shnaidman E. Putative role of adipose tissue in growth and metabolism of colon cancer cells. Front Oncol. 2014;4:1–7.
Yehuda-Shnaidman E, Nimri L, Tarnovscki T, Kirshtein B, Rudich A, Schwartz B. Secreted human adipose leptin decreases mitochondrial respiration in HCT116 colon cancer cells. Bermano G, editor. PLoS ONE. 2013;8:e74843.
Antoun S, Bayar A, Ileana E, Laplanche A, Fizazi K, di Palma M, et al. High subcutaneous adipose tissue predicts the prognosis in metastatic castration-resistant prostate cancer patients in post chemotherapy setting. Eur J Cancer. 2015;51:2570–7.
Ebadi M, Martin L, Ghosh S, Field CJ, Lehner R, Baracos VE, et al. Subcutaneous adiposity is an independent predictor of mortality in cancer patients. Br J Cancer. 2017;117:148–55.
Aleksandrova K, Mozaffarian D, Pischon T. Addressing the perfect storm: biomarkers in obesity and pathophysiology of cardiometabolic risk. Clin Chem. 2018;64:142–53.
Benito PJ, Gómez-Candela C, Cabañas MD, Szendrei B, Castro EA. Comparison between different methods for measuring body fat after a weight loss program. Rev Bras Med do Esporte. 2019;25:474–9.
Allott EH, Hursting SD. Obesity and cancer: mechanistic insights from transdisciplinary studies. Endocr Relat Cancer. 2015;22:R365–86.
Coelho M, Oliveira T, Fernandes R. State of the art paper Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;2:191–200.
Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–56.
Booth A, Magnuson A, Fouts J, Foster M. Adipose tissue, obesity and adipokines: role in cancer promotion. Horm Mol Biol Clin Invest. 2015;21:57–74.
Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013;1831:1533–41.
Dong Z, Fu S, Xu X, Yang Y, Du L, Li W, et al. Leptin-mediated regulation of ICAM-1 is Rho/ROCK dependent and enhances gastric cancer cell migration. Br J Cancer. 2014;110:1801–10.
Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-associated adipocytes: key players in breast cancer progression. J Hematol Oncol. 2019;12:95.
Bernard JJ, Wellberg EA. The tumor promotional role of adipocytes in the breast cancer micro- and macro-environment. Am J Pathol. 2021;723:138028.
Li S, Wu T, Lu Y-X, Wang J-X, Yu F-H, Yang M-Z, et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020;36:101596.
Gallo M, Adinolfi V, Barucca V, Prinzi N, Renzelli V, Barrea L, et al. Expected and paradoxical effects of obesity on cancer treatment response. Rev Endocr Metab Disord. 2020. https://doi.org/10.1007/s11154-020-09597-y.
Hotamisligil G, Shargill N, Spiegelman B. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32.
Muoio DM, Dohm GL, Tapscott EB, Coleman RA. Leptin opposes insulin’s effects on fatty acid partitioning in muscles isolated from obese ob/ob mice. Am J Physiol Metab. 1999;276:E913–21.
Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol. 1999;194:6–11.
Martín-Romero C, Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell Immunol. 2000;199:15–24.
Chandra RK, Au B. Spleen hemolytic plaque-forming cell response and generation of cytotoxic cells in genetically obese (C57B1/6J ob/ob) mice. Int Arch Allergy Immunol. 1980;62:94–8.
Fernandes G, Handwerger BS, Yunis EJ, Brown DM. Immune response in the mutant diabetic C57BL/Ks-dt+ mouse. Discrepancies between in vitro and in vivo immunological assays. J Clin Invest. 1978;61:243–50.
Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation- induced immunosuppression. Nature. 1998;394:897–901.
Pérez-Pérez A, Vilariño-García T, Fernández-Riejos P, Martín-González J, Segura-Egea JJ, Sánchez-Margalet V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017;35:71–84.
Considine R V., Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–5. https://doi.org/10.1056/NEJM199602013340503.
Lönnqvist F, Arner P, Nordfors L, Schalling M. Overexpression of the obese (ob) gene in adipose tissue of human obese subjects. Nat Med. 1995;1:950–3.
Soliman AT, ElZalabany MM, Salama M, Ansari BM. Serum leptin concentrations during severe protein-energy malnutrition: correlation with growth parameters and endocrine function. Metabolism. 2000;49:819–25.
Palacio A, Lopez M, Perez-Bravo F, Monkeberg F, Schlesinger L. Leptin levels are associated with immune response in malnourished infants. J Clin Endocrinol Metab. 2002;87:3040–6. https://doi.org/10.1210/jcem.87.7.8636.
Jiang L, Su H, Wu X, Shen H, Kim M-H, Li Y, et al. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat Commun. 2020;11:1517.
Chrysafi P, Perakakis N, Farr OM, Stefanakis K, Peradze N, Sala-Vila A, et al. Leptin alters energy intake and fat mass but not energy expenditure in lean subjects. Nat Commun. 2020;11:5145.
Vona-Davis L, Rose DP. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr Relat Cancer. 2007;14:189–206.
Jiménez-Cortegana C, López-Saavedra A, Sánchez-Jiménez F, Pérez-Pérez A, Castiñeiras J, Virizuela-Echaburu JA, et al. Leptin, both bad and good actor in cancer. Biomolecules. 2021;11:913.
Jardé T, Caldefie-Chézet F, Goncalves-Mendes N, Mishellany F, Buechler C, Penault-Llorca F, et al. Involvement of adiponectin and leptin in breast cancer: clinical and in vitro studies. Endocr Relat Cancer. 2009;16:1197–210.
García-Robles MJ, Segura-Ortega JE, Fafutis-Morris M. The biology of leptin and its implications in breast cancer: a general view. J Interf Cytokine Res. 2013;33:717–27.
Jardé T, Perrier S, Vasson M-P, Caldefie-Chézet F. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer. 2011;47:33–43.
Zheng Q, Dunlap SM, Zhu J, Downs-Kelly E, Rich J, Hursting SD, et al. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr Relat Cancer. 2011;18:491–503.
Zheng Q, Banaszak L, Fracci S, Basali D, Dunlap SM, Hursting SD, et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr Relat Cancer. 2013;20:797–808.
Xiong Y, Zhang J, Liu M, An M, Lei L, Guo W. Human leptin protein activates the growth of HepG2 cells by inhibiting PERK-mediated ER stress and apoptosis. Mol Med Rep. 2014;10:1649–55.
Kim JH, Lee SY, Myung SC, Kim YS, Kim T-H, Kim MK. Clinical significance of the leptin and leptin receptor expressions in prostate tissues. Asian J Androl. 2008;10:923–8.
Tewari R, Rajender S, Natu SM, Goel A, Dalela D, Goel MM, et al. Significance of obesity markers and adipocytokines in high grade and high stage prostate cancer in North Indian men—a cross-sectional study. Cytokine. 2013;63:130–4.
Bain GH, Collie-Duguid E, Murray GI, Gilbert FJ, Denison A, Mckiddie F, et al. Tumour expression of leptin is associated with chemotherapy resistance and therapy-independent prognosis in gastro-oesophageal adenocarcinomas. Br J Cancer. 2014;110:1525–34.
von Frankenberg AD, Reis AF, Gerchman F. Relationships between adiponectin levels, the metabolic syndrome, and type 2 diabetes: a literature review. Arch Endocrinol Metab. 2017;61:614–22.
Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–51.
Barb D, Williams CJ, Neuwirth AK, Mantzoros CS. Adiponectin in relation to malignancies: a review of existing basic research and clinical evidence. Am J Clin Nutr. 2007;86:858S-866S.
Katira A, Tan PH. Evolving role of adiponectin in cancer-controversies and update. Cancer Biol Med. 2016;13:101–19.
Lu W, Huang Z, Li N, Liu H. Low circulating total adiponectin, especially its non-high-molecular weight fraction, represents a promising risk factor for colorectal cancer: a meta-analysis. OncoTargets Ther. 2018;11:2519–31.
Yoon YS, Kwon AR, Lee YK, Oh SW. Circulating adipokines and risk of obesity related cancers: a systematic review and meta-analysis. Obes Res Clin Pract. 2019;13:329–39.
Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci. 2017;18:1321.
Yamauchi N, Takazawa Y, Maeda D, Hibiya T, Tanaka M, Iwabu M, et al. Expression levels of adiponectin receptors are decreased in human endometrial adenocarcinoma tissues. Int J Gynecol Pathol. 2012;31:352–7.
Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab. 2008;93:s64-73.
Suraj J, Kurpińska A, Zakrzewska A, Sternak M, Stojak M, Jasztal A, et al. Early and late endothelial response in breast cancer metastasis in mice: simultaneous quantification of endothelial biomarkers using a mass spectrometry-based method. Dis Model Mech. 2019;12:dmm036269.
Dieudonne M-N, Bussiere M, Dos Santos E, Leneveu M-C, Giudicelli Y, Pecquery R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem Biophys Res Commun. 2006;345:271–9.
Erdogan S, Sezer S, Baser E, Gun-Eryilmaz O, Gungor T, Uysal S, et al. Evaluating vaspin and adiponectin in postmenopausal women with endometrial cancer. Endocr Relat Cancer. 2013;20:669–75.
Goktas S, Yilmaz MI, Caglar K, Sonmez A, Kilic S, Bedir S. Prostate cancer and adiponectin. Urology. 2005;65:1168–72.
Nakajima TE, Yamada Y, Hamano T, Furuta K, Matsuda T, Fujita S, et al. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci. 2010;101:1286–91.
Alexandre L. Pathophysiological mechanisms linking obesity and esophageal adenocarcinoma. World J Gastrointest Pathophysiol. 2014;5:534.
Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, inflammation, and cancer. Annu Rev Pathol Mech Dis. 2016;11:421–49.
Boutari C, Mantzoros CS. Inflammation: a key player linking obesity with malignancies. Metabolism. 2018;81:A3–6.
Wang L, Zhao R, Song X, Wu W. Targeting ERβ in macrophage reduces crown-like structures in adipose tissue by inhibiting osteopontin and HIF-1α. Sci Rep. 2019;9:15762.
Unamuno X, Gómez-Ambrosi J, Rodríguez A, Becerril S, Frühbeck G, Catalán V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur J Clin Invest. 2018;48:e12997.
Ferrante AW. The immune cells in adipose tissue. Diabetes Obes Metab. 2013;15:34–8.
Tsuhako R, Yoshida H, Sugita C, Kurokawa M. Naringenin suppresses neutrophil infiltration into adipose tissue in high-fat diet-induced obese mice. J Nat Med. 2020;74:229–37.
Chylikova J, Dvorackova J, Tauber Z, Kamarad V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed Pap. 2018;162:79–82.
Wang Q, Wu H. T cells in adipose tissue: critical players in immunometabolism. Front Immunol. 2018;9:2509.
Divella R, De Luca R, Abbate I, Naglieri E, Daniele A. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J Cancer. 2016;7:2346–59.
Mraz M, Haluzik M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol. 2014;222:R113–27.
Asghar A, Sheikh N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell Immunol. 2017;315:18–26.
Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes. Arterioscler Thromb Vasc Biol. 2005;25:2062–8.
Martins LM, Oliveira ARS, Cruz KJC, Torres-Leal FL, Marreiro D do N. Obesity, inflammation, and insulin resistance. Braz J Pharm Sci. 2014;50:677–92.
Hivert M-F, Sullivan LM, Fox CS, Nathan DM, D’Agostino RB, Wilson PWF, et al. Associations of adiponectin, resistin, and tumor necrosis factor-α with insulin resistance. J Clin Endocrinol Metab. 2008;93:3165–72.
Gao D, Madi M, Ding C, Fok M, Steele T, Ford C, et al. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am J Physiol Metab. 2014;307:E289-304.
Baker KJ, Houston A, Brint E. IL-1 family members in cancer; two sides to every story. Front Immunol. 2019;10:1197.
Cui G, Yuan A, Sun Z, Zheng W, Pang Z. IL-1β/IL-6 network in the tumor microenvironment of human colorectal cancer. Pathol Res Pract. 2018;214:986–92.
Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–65.
Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–21.
Ross KH, Gogineni K, Subhedar PD, Lin JY, McCullough LE. Obesity and cancer treatment efficacy: existing challenges and opportunities. Cancer. 2019;125:1588–92.
Amri R, Bordeianou LG, Sylla P, Berger DL. Obesity, outcomes and quality of care: body mass index increases the risk of wound-related complications in colon cancer surgery. Am J Surg. 2014;207:17–23.
de Blacam C, Ogunleye AA, Momoh AO, Colakoglu S, Tobias AM, Sharma R, et al. High body mass index and smoking predict morbidity in breast cancer surgery. Ann Surg. 2012;255:551–5.
Ramsey AM, Martin RC. Body mass index and outcomes from pancreatic resection: a review and meta-analysis. J Gastrointest Surg. 2011;15:1633–42.
Mullen JT, Davenport DL, Hutter MM, Hosokawa PW, Henderson WG, Khuri SF, et al. Impact of body mass index on perioperative outcomes in patients undergoing major intra-abdominal cancer surgery. Ann Surg Oncol. 2008;15:2164–72.
Tokunaga M, Hiki N, Fukunaga T, Ohyama S, Yamaguchi T, Nakajima T. Better 5-year survival rate following curative gastrectomy in overweight patients. Ann Surg Oncol. 2009;16:3245–51.
Tsai S, Choti MA, Assumpcao L, Cameron JL, Gleisner AL, Herman JM, et al. Impact of obesity on perioperative outcomes and survival following pancreaticoduodenectomy for pancreatic cancer: a large single-institution study. J Gastrointest Surg. 2010;14:1143–50.
Li S, Che G, Liu L, Chen L. Does the “obesity paradox” really exist in lung cancer surgery?—maybe we should recognize what is the “obesity” first. J Thorac Dis. 2019;11:S291–5.
Wang LS, Murphy CT, Ruth K, Zaorsky NG, Smaldone MC, Sobczak ML, et al. Impact of obesity on outcomes after definitive dose-escalated intensity-modulated radiotherapy for localized prostate cancer. Cancer. 2015;121:3010–7.
Gross JP, Strauss JB, Lurain J, Berry E, Neubauer N, Helenowski I, et al. Impact of obesity on treatment-related adverse events, disease recurrence, and survival in women with cervical carcinoma. J Radiat Oncol. 2016;5:197–203.
De Langhe S, Mulliez T, Veldeman L, Remouchamps V, van Greveling A, Gilsoul M, et al. Factors modifying the risk for developing acute skin toxicity after whole-breast intensity modulated radiotherapy. BMC Cancer. 2014;14:711.
Goldsmith C, Haviland J, Tsang Y, Sydenham M, Yarnold J. Large breast size as a risk factor for late adverse effects of breast radiotherapy: is residual dose inhomogeneity, despite 3D treatment planning and delivery, the main explanation? Radiother Oncol. 2011;100:236–40.
Litton JK, Gonzalez-Angulo AM, Warneke CL, Buzdar AU, Kau S-W, Bondy M, et al. Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. J Clin Oncol. 2008;26:4072–7.
Lashinger LM, Rossi EL, Hursting SD. Obesity and resistance to cancer chemotherapy: interacting roles of inflammation and metabolic dysregulation. Clin Pharmacol Ther. 2014;96:458–63.
Wong AL, Seng KY, Ong EM, Wang LZ, Oscar H, Cordero MT, et al. Body fat composition impacts the hematologic toxicities and pharmacokinetics of doxorubicin in Asian breast cancer patients. Breast Cancer Res Treat. 2014;144:143–52.
Hourdequin KC, Schpero WL, McKenna DR, Piazik BL, Larson RJ. Toxic effect of chemotherapy dosing using actual body weight in obese versus normal-weight patients: a systematic review and meta-analysis. Ann Oncol. 2013;24:2952–62.
Georgiadis MS, Steinberg SM, Hankins LA, Ihde DC, Johnson BE. Obesity and therapy-related toxicity in patients treated for small-cell lung cancer. J Natl Cancer Inst. 1995;87:361–6.
Jenkins P, Elyan S, Freeman S. Obesity is not associated with increased myelosuppression in patients receiving chemotherapy for breast cancer. Eur J Cancer. 2007;43:544–8.
Wright JD, Tian C, Mutch DG, Herzog TJ, Nagao S, Fujiwara K, et al. Carboplatin dosing in obese women with ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109:353–8.
Lehuédé C, Li X, Dauvillier S, Vaysse C, Franchet C, Clement E, et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: role of the major vault protein (MVP). Breast Cancer Res. 2019;21:7.
Bowers L, Kulkarni A, Hursting S. Obesity-associated leptin signaling promotes chemotherapy resistance in basal-like breast cancer: the role of tumor-associated macrophages. Curr Dev Nutr. 2020;4:311–311.
Cespedes Feliciano EM, Kroenke CH, Caan BJ. The obesity paradox in cancer: how important is muscle? Annu Rev Nutr. 2018;38:357–79.
Griggs JJ, Mangu PB, Temin S, Lyman GH. Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Oncol Pract. 2012;8:e59-61.
Lyman GH, Sparreboom A. Chemotherapy dosing in overweight and obese patients with cancer. Nat Rev Clin Oncol. 2013;10:451–9.
Kalantar-Zadeh K, Rhee CM, Chou J, Ahmadi SF, Park J, Chen JLT, et al. The obesity paradox in kidney disease: how to reconcile it with obesity management. Kidney Int Rep. 2017;2:271–81.
Horowitz NS, Wright AA. Impact of obesity on chemotherapy management and outcomes in women with gynecologic malignancies. Gynecol Oncol. 2015;138:201–6.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Oiseth SJ, Aziz MS. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat. 2017;3:250.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–68.
Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13:1–15.
Yang JC, Rosenberg SA. Adoptive T-cell therapy for cancer. Adv Immunol. 2016;130:279–94.
Sadeghzadeh M, Bornehdeli S, Mohahammadrezakhani H, Abolghasemi M, Poursaei E, Asadi M, et al. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020;254:117580.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Rezvani K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant. 2019;54:785–8.
Lee KL, Schlom J, Hamilton DH. Combination therapies utilizing neoepitope-targeted vaccines. Cancer Immunol Immunother. 2021;70:875–85.
Rogers LM, Veeramani S, Weiner GJ. Complement in monoclonal antibody therapy of cancer. Immunol Res. 2014;59:203–10.
Fritz JM, Lenardo MJ. Development of immune checkpoint therapy for cancer. J Exp Med. 2019;216:1244–54.
Barros L, Pretti MA, Chicaybam L, Abdo L, Boroni M, Bonamino MH. Immunological-based approaches for cancer therapy. Clinics. 2018;73:1–11.
Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol. 2020;13:100738.
Marcucci F, Rumio C, Corti A. Tumor cell-associated immune checkpoint molecules—drivers of malignancy and stemness. Biochim Biophys Acta. 2017;1868:571–83.
Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett. 2017;192:1–6.
Vigneron N. Human tumor antigens and cancer immunotherapy. Biomed Res Int. 2015. https://doi.org/10.1155/2015/948501.
Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96.
Palucka AK, Coussens LM. The basis of oncoimmunology. Cell. 2016;164:1233–47.
Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32:1267–84.
Ardolino L, Joshua A. Immune checkpoint inhibitors in malignancy. Aust Prescr. 2019;42:62.
Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–86.
Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med. 2018;283:110–20.
Ujvari B, Jacqueline C, Misse D, Amar V, Fitzpatrick JC, Jennings G, et al. Obesity paradox in cancer: is bigger really better? Evol Appl. 2019;12:1092–5.
Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33:570–80.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science. 2018;359:1366–70.
Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14:356–65.
Surmi BK, Hasty AH. Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol. 2008;3:545–56.
Woodall MJ, Neumann S, Campbell K, Pattison ST, Young SL. The effects of obesity on anti-cancer immunity and cancer immunotherapy. Cancers (Basel). 2020;12:1–14.
Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, MacIver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 2014;192:136–44.
Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med. 2019;25:141–51.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38.
Tsang NM, Pai PC, Chuang CC, Chuang WC, Tseng CK, Chang KP, et al. Overweight and obesity predict better overall survival rates in cancer patients with distant metastases. Cancer Med. 2016;5:665–75.
Albiges L, Ari Hakimi A, Xie W, McKay RR, Simantov R, Lin X, et al. Body mass index and metastatic renal cell carcinoma: clinical and biological correlations. J Clin Oncol. 2016;34:3655–63.
Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AWK, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9.
Clements VK, Long T, Long R, Figley C, Smith DMC, Ostrand-Rosenberg S. Frontline science: high fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J Leukoc Biol. 2018;103:395–407.
Yang H, Youm Y-H, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood. 2009;114:3803–12.
Corrêa LH, Corrêa R, Farinasso CM, de Sant’Ana Dourado LP, Magalhães KG. Adipocytes and macrophages interplay in the orchestration of tumor microenvironment: new implications in cancer progression. Front Immunol. 2017;8:1129.
Murphy WJ, Longo DL. The surprisingly positive association between obesity and cancer immunotherapy efficacy. JAMA. 2019;321:1247.
Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med. 2019;25:141–51.
Cortellini A, Bersanelli M, Buti S, Cannita K, Santini D, Perrone F, et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: when overweight becomes favorable. J Immunother Cancer. 2019;7:57.
Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7:410–20.
Zeeni N, Dagher-Hamalian C, Dimassi H, Faour WH. Cafeteria diet-fed mice is a pertinent model of obesity-induced organ damage: a potential role of inflammation. Inflamm Res. 2015;64:501–12. https://doi.org/10.1007/s00011-015-0831-z.
Lang P, Hasselwander S, Li H, Xia N. Effects of different diets used in diet-induced obesity models on insulin resistance and vascular dysfunction in C57BL/6 mice. Sci Rep. 2019;9:19556.
Sumiyoshi M, Sakanaka M, Kimura Y. Chronic intake of high-fat and high-sucrose diets differentially affects glucose intolerance in mice. J Nutr. 2006;136:582–7.
Murphy KA, James BR, Sjaastad FV, Kucaba TA, Kim H, Brincks EL, et al. Cutting edge: elevated leptin during diet-induced obesity reduces the efficacy of tumor immunotherapy. J Immunol. 2018;201:1837–41.
Cranford TL, Velázquez KT, Enos RT, Sougiannis AT, Bader JE, Carson MS, et al. Effects of high fat diet-induced obesity on mammary tumorigenesis in the PyMT/MMTV murine model. Cancer Biol Ther. 2019;20:487–96. https://doi.org/10.1080/15384047.2018.1537574.
Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. Mattei F, editor. PLoS ONE. 2016;11:e0157164. https://doi.org/10.1371/journal.pone.0157164.
Gibson JT, Orlandella RM, Turbitt WJ, Behring M, Manne U, Sorge RE, et al. Obesity-associated myeloid-derived suppressor cells promote apoptosis of tumor-infiltrating CD8 T cells and immunotherapy resistance in breast cancer. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.590794/full.
Daniel H, Gholami AM, Berry D, Desmarchelier C, Hahne H, Loh G, et al. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014;8:295–308.
de Sant’Ana LP, Ribeiro DJS, Martins AMA, dos Santos FN, Corrêa R, Almeida R das N, et al. Absence of the caspases 1/11 modulates liver global lipid profile and gut microbiota in high-fat-diet-induced obese mice. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2019.02926/full.
Schulz MD, Atay Ç, Heringer J, Romrig FK, Schwitalla S, Aydin B, et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature. 2014;514:508–12.
Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24:1804–8.
Baiden-Amissah REM, Tuyaerts S. Contribution of aging, obesity, and microbiota on tumor immunotherapy efficacy and toxicity. Int J Mol Sci. 2019;20:3586.
McQuade JL, Daniel CR, Helmink BA, Wargo JA. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019;20:e77–91.
Naik GS, Waikar SS, Johnson AEW, Buchbinder EI, Haq R, Hodi FS, et al. Complex inter-relationship of body mass index, gender and serum creatinine on survival: exploring the obesity paradox in melanoma patients treated with checkpoint inhibition. J Immunother Cancer. 2019;7:89.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381:1535–46.
Smith LK, Arabi S, Lelliott EJ, McArthur GA, Sheppard KE. Obesity and the impact on cutaneous melanoma: friend or foe? Cancers (Basel). 2020;12:1583.
Richtig G, Hoeller C, Wolf M, Wolf I, Rainer BM, Schulter G, et al. Body mass index may predict the response to ipilimumab in metastatic melanoma: an observational multi-centre study. Haass NK, editor. PLoS ONE. 2018;13:e0204729.
Chinai JM, Janakiram M, Chen F, Chen W, Kaplan M, Zang X. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol Sci. 2015;36:587–95.
Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernández C, Mera A, Gonzalez-Gay MA, et al. Obesity, fat mass and immune system: role for leptin. Front Physiol. 2018;9:1–20.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16:626–38.
Joosse A, Collette S, Suciu S, Nijsten T, Lejeune F, Kleeberg UR, et al. Superior outcome of women with stage I/II cutaneous melanoma: pooled analysis of four European organisation for research and treatment of cancer phase III trials. J Clin Oncol. 2012;30:2240–7.
Fang S, Wang Y, Dang Y, Gagel A, Ross MI, Gershenwald JE, et al. Association between body mass index, C-reactive protein levels, and melanoma patient outcomes. J Invest Dermatol. 2017;137:1792–5.
Natale CA, Duperret EK, Zhang J, Sadeghi R, Dahal A, O’Brien KT, et al. Sex steroids regulate skin pigmentation through nonclassical membrane-bound receptors. eLife. 2016;5:1–16.
Natale CA, Li J, Zhang J, Dahal A, Dentchev T, Stanger BZ, et al. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. eLife. 2018;7:1–19.
Indini A, Rijavec E, Ghidini M, Tomasello G, Cattaneo M, Barbin F, et al. Impact of BMI on survival outcomes of immunotherapy in solid tumors: a systematic review. Int J Mol Sci. 2021;22:2628.
Cortellini A, Ricciuti B, Tiseo M, Bria E, Banna GL, Aerts JGJV, et al. Baseline BMI and BMI variation during first line pembrolizumab in NSCLC patients with a PD-L1 expression ≥ 50%: a multicenter study with external validation. J Immunother Cancer. 2020;8:e001403.
Ichihara E, Harada D, Inoue K, Sato K, Hosokawa S, Kishino D, et al. The impact of body mass index on the efficacy of anti-PD-1/PD-L1 antibodies in patients with non-small cell lung cancer. Lung Cancer. 2020;139:140–5.
Popinat G, Cousse S, Goldfarb L, Becker S, Gardin I, Salaün M, et al. Sub-cutaneous fat mass measured on multislice computed tomography of pretreatment PET/CT is a prognostic factor of stage IV non-small cell lung cancer treated by nivolumab. OncoImmunology. 2019;8:e1580128.
Kichenadasse G, Miners JO, Mangoni AA, Rowland A, Hopkins AM, Sorich MJ. Association between body mass index and overall survival with immune checkpoint inhibitor therapy for advanced non-small cell lung cancer. JAMA Oncol. 2020;6:512.
Magri V, Gottfried T, Di Segni M, Urban D, Peled M, Daher S, et al. Correlation of body composition by computerized tomography and metabolic parameters with survival of nivolumab-treated lung cancer patients. Cancer Manag Res. 2019;11:8201–7.
Li M, Bu R. Biological support to obesity paradox in renal cell carcinoma: a review. Urol Int. 2020;104:837–48.
De Giorgi U, Procopio G, Giannarelli Di, Sabbatini R, Bearz A, Buti S, et al. Association of systemic inflammation index and body mass index with survival in patients with renal cell cancer treated with nivolumab. Clin Cancer Res. 2019;25:3839–46.
Martini DJ, Liu Y, Shabto JM, Carthon BC, Hitron EE, Russler GA, et al. Novel risk scoring system for patients with metastatic renal cell carcinoma treated with immune checkpoint inhibitors. Oncologist. 2020;25:484–91.
Labadie BW, Liu P, Bao R, Crist M, Fernandes R, Ferreira L, et al. BMI, irAE, and gene expression signatures associate with resistance to immune-checkpoint inhibition and outcomes in renal cell carcinoma. J Transl Med. 2019;17:386.
Boi SK, Orlandella RM, Gibson JT, Turbitt WJ, Wald G, Thomas L, et al. Obesity diminishes response to PD-1-based immunotherapies in renal cancer. J Immunother Cancer. 2020;8:e000725.
Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, et al. Adipose PD-L1 modulates PD-1/PD-L1 checkpoint blockade immunotherapy efficacy in breast cancer. OncoImmunology. 2018;7:e1500107.
Wu B, Chiang H-C, Sun X, Yuan B, Mitra P, Hu Y, et al. Genetic ablation of adipocyte PD-L1 reduces tumor growth but accentuates obesity-associated inflammation. J Immunother Cancer. 2020;8:e000964.
Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, et al. Adipocyte PD-L1 suppresses anti-tumor immune response and promotes breast cancer progression. J Immunol. 2020;204:165.24.
Wang X, Qi Y, Kong X, Zhai J, Li Y, Song Y, et al. Immunological therapy: a novel thriving area for triple-negative breast cancer treatment. Cancer Lett. 2019;442:409–28.
Liu Y-H, Zang X-Y, Wang J-C, Huang S-S, Xu J, Zhang P. Diagnosis and management of immune related adverse events (irAEs) in cancer immunotherapy. Biomed Pharmacother. 2019;120:109437.
Mirsoian A, Bouchlaka MN, Sckisel GD, Chen M, Pai CCS, Maverakis E, et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J Exp Med. 2014;211:2373–83.
Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45:e124–31.
Li X, Shao C, Shi Y, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol Oncol. 2018;11:31.
Yuan Y, Adam A, Zhao C, Chen H. Recent advancements in the mechanisms underlying resistance to PD-1/PD-L1 blockade immunotherapy. Cancers (Basel). 2021;13:663.
Eun Y, Kim IY, Kim H, Ahn JK, Park E-J, Hwang J, et al. SAT0715 risk factors of immune-related adverse events in patients treated with anti-pd-1 antibody pembrolizumab. Saturday, 16 JUNE 2018. BMJ Publishing Group Ltd and European League Against Rheumatism; 2018. p. 1205.
Rogado J, Romero-Laorden N, Sanchez-Torres JM, Ramos-Levi AM, Pacheco-Barcia V, Ballesteros AI, et al. Effect of excess weight and immune-related adverse events on the efficacy of cancer immunotherapy with anti-PD-1 antibodies. OncoImmunology. 2020;9:1751548.
Acknowledgements
None.
Funding
This work was supported by CNPq - National Council for Scientific and Technological Development (grant # 313106/2020-7 and 311761/2018-6), FAPERJ - Foundation for Research Support of the State of Rio de Janeiro (grant # CNE-E-26/202.645/2019) and Ministry of Health (Brazil).
Author information
Authors and Affiliations
Contributions
JAFS, GP, and MVD wrote different sections of the manuscript. JAFS prepared the figures. MHB revised and edited the manuscript. KGM prepared, revised, and wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Assumpção, J.A.F., Pasquarelli-do-Nascimento, G., Duarte, M.S.V. et al. The ambiguous role of obesity in oncology by promoting cancer but boosting antitumor immunotherapy. J Biomed Sci 29, 12 (2022). https://doi.org/10.1186/s12929-022-00796-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-022-00796-0