
Abstract
Epigenetic drug discovery field has evidenced significant advancement in the recent times. A plethora of small molecule inhibitors have progressed to clinical stage investigations and are being explored exhaustively to ascertain conclusive benefits in diverse malignancies. Literature precedents indicates that substantial amount of efforts were directed towards the use of epigenetic tools in monotherapy as well as in combination regimens at the clinical level, however, the preclinical/preliminary explorations were inclined towards the identification of prudent approaches that can leverage the anticancer potential of small molecule epigenetic inhibitors as single agents only. This review article presents an update of FDA approved epigenetic drugs along with the epigenetic inhibitors undergoing clinical stage investigations in different cancer types. A detailed discussion of the pragmatic strategies that are expected to steer the progress of the epigenetic therapy through the implementation of emerging approaches such as PROTACS and CRISPR/Cas9 along with logical ways for scaffold fabrication to selectively approach the enzyme isoforms in pursuit of garnering amplified antitumor effects has been covered. In addition, the compilation also presents the rational strategies for the construction of multi-targeting scaffold assemblages employing previously identified pharmacophores as potential alternatives to the combination therapy.
Similar content being viewed by others

Background
Genome refers to the complete set of genetic information in the form of nucleotide sequence inside the DNA, whereas the epigenome refers to complex modifications inside the genomic DNA [1]. In simple terms, epigenetics involves a set of structural modifications within the nucleic acids and histone that do not involve a change in an individual’s genetic code [2,3,4] and can be termed as ‘on top’ or ‘in addition’ to genetics [5].
Epigenetic mechanisms regulate gene transcription and genomic stability and maintain normal cell growth, development, and differentiation [6,7,8,9,10]. As such, epigenetic regulation is a dynamic and reversible process and epigenetic modifications are carried out by writers (DNMTs, HATs, ubiquitin E3 ligases and HMTs) that catalyze the addition of epigenetic marks onto either DNA or histone tails, readers (bromodomains) that recognizes or are recruited to a specific epigenetic mark and erasers (HDACs, KDMs and deubiquitinating enzymes) that removes the epigenetic marks [11,12,13,14,15,16,17,18,19,20,21].
Though epigenetics is a key component of an organism’s normal development, from embryonic development through adulthood, epigenetic dysregulation can significantly contribute to the origin and progression of human diseases such as cancer, cardiovascular diseases, metabolic diseases and neurological diseases. Extensive explorations conducted to enhance the understanding of the epigenome reveals that localised differences existing in epigenetic states of normal and disease tissues can be utilized as disease biomarkers [22,23,24,25,26].
Literature precedents indicate that all the three families of epigenetic proteins—readers, writers, and erasers are druggable targets. This disclosure coupled with the improved understanding of epigenetics in diverse complications dramatically spurred and expedited the translational investigation of the epigenetic inhibitors. In particular, exhaustive investigations predominantly on small-molecule inhibitors were carried out at the clinical level and the subsequent efforts have culminated in the identification of efficacious inhibitors, with some of them being used in the clinic currently. To add on, the preclinical and preliminary studies have also comprehensively explored the epigenetic tools (DNMT/HDAC/LSD1/DOT1L/BET/EZH2 inhibitors) in pursuit of leveraging enhanced antiproliferative effects. Albeit the clinical stage investigations have been appropriately directed towards the evaluation of the epigenetic inhibitors as single agents, a significant proportion of the efforts is also covered by the studies inclined towards the utilization of the epigenetic tools as a part of combination regimens. This information raises a critical question regarding the therapeutic credibility of some of the epigenetic inhibitors as single agents to attain conclusive benefits in cancer. The doubts are further strengthened by the fact that only seven drugs have approved till date despite the epigenetic targets being at the forefront of the strategized explorations. Nevertheless, the medicinal chemist at the preclinical/preliminary level has been quite proficient to employ rational drug design approaches to maximize the benefits of the predefined pharmacophore models of the epigenetic targets. Indeed, the preclinical/preliminary findings (section) bears a relatively higher degree of fascination for the researchers as efforts invested have not just been confined to elucidate the mechanistic insights responsible for exerting antitumor effects via inhibition of the epigenetic targets, rather the chemist has looked beyond this strategy to attain favourable effects via degradation of the proteins also (PROTACS). Sagaciously evidenced on the literature precedential basis, degradation of the target proteins can be achieved at low exposures by PROTACs (protein degraders) owing to their catalytic mode of action and this emerging approach is likely to steer the wheels of the drug discovery field towards the class of degraders bearing appropriately installed epigenetic tools in the near future. For the selective targeting, the concept of antibody–drug conjugates have also attracted the eyeballs of the researchers working in the field of epigenetic inhibitors. This strategy of targeted drug delivery is anticipated to overcome the issue of systemic toxicity and narrow therapeutic window that limits the clinical use of the available epigenetic inhibitors. CRISPR/Cas9-based strategies to target the cancerous epigenetic regulators also represent an emerging potential approach that is being foreseen as a tool to correct genetic mutations. Other than this, the approach of multitarget assemblage construction has continued garnering significant attention to extract enhanced antitumor effects via concomitant inhibition of the biochemically correlated targets and is also conceived to be one of the preferred futuristic strategies as a potential alternative to the combination therapy. To sum up, it is highly likely that the ship of epigenetic inhibitors will sail through the implementation of the aforementioned approaches.
Despite the significant promise demonstrated by the aforementioned strategies, there is no denying the fact that the conventional approaches will continue receiving tantamount attention of the research groups for the development of new inhibitors. Fragment stitching approach on existing drugs coupled with lead modification studies ascertaining the impact of scaffold installation, regiovariation, bioisosteric replacement, structure simplification approach, structure rigidification approach and other subtle structural variations on the activity profile exemplifies some of these potential approaches.
In light of the current scenario and the amount of efforts currently being invested in this field, it is highly likely that this decade might evidence the therapeutic growth of a handful of epigenetic drugs presently undergoing efficacy and safety evaluations at the clinical level and many new agents might enter the clinic. This review article presents an update of FDA approved epigenetic drugs along with the epigenetic inhibitors undergoing clinical stage investigations. The compilation also encompasses a detailed discussion of the rational strategies that can prove to be instrumental in the development of new inhibitors. The covered literature in this review indicates that the future attempts in the epigenetic drug discovery filed needs to headed in the following directions: (i) explorations of natural product based libraries for the development of non-nucleoside based DNMT inhibitors (ii) initiation of parallel programs on non-metal chelating type HDAC inhibitors as well as anilides to transpose the focus from hydroxamic acid type scaffolds owing to the pharmacological liabilities associated with latter class (iii) exhaustive studies needs to be conducted to ascertain the expression level of epigenetic enzymes in diverse malignancies (iv) fabrication of selective isoform inhibitors of HDAC to extract amplified anticancer effects despite of the fact that the clinical success, till date, have only been attained through pan HDAC inhibitors (v) exploration of additional structural templates other than the framework of tranylcypromine to expand the size of LSD1 inhibitors pipeline (vi) design of dual EZH1/EZH2 inhibitors in view of the fact EZH1, complements EZH2 in mediating H3K27 methylation and is also endowed with HMT activity. (vii) Expanding the size of the libraries of DOT1L inhibitors (viii) utilization of the existing chemical architectures of BET and HDAC inhibitors in the PROTAC model and antibody–drug conjugate model (ix) explorations of combination of epigenetic inhibitors with immunotherapy.
Epigenetics and cancer
Epigenetic processes comprises of inherited, somatic and reversible changes in gene expression in cancer cells. DNA methylation, histone modification (acetylation, methylation, phosphorylation, etc.) and noncoding RNAs are the major epigenetic mechanisms that control gene activity leading to a number of complex cancers [4]. In most of the cancers, DNA is hypomethylated along with the hypermethylation at other sites [27]. The two anomalous processes i.e. hypomethylation and hypermethylation activates oncogenes and inhibits the tumor suppressor genes, respectively [28]. Apart from methylation process, histone modification is another process that plays important role in cancer. Histone modifications control the active and inactive state of chromatin which ultimately influences the gene expression within the former region [29]. MicroRNAs are responsible for degradation of mRNA as well as inhibition of target mRNA through respective complementary base pairing and partial base pairing [30]. All these epigenetic changes start taking place a long time ago before the occurrence of cancer and are considered accountable for any genetic changes in cancer, also labelling them as “first hits” for tumorigeneses [27].
Role of DNA methylation in cancer cells
DNA methylation is an epigenetic process that can be described as the covalent transfer of methyl groups to the fifth carbon of cytosine (5-mC) within 5′-CpG-3′ dinucleotides catalysed by DNMTs with SAM as the methyl donor [31, 32]. In mammals, three major types of DNMT enzymes are found, DNMT1, DNMT3a, and DNMT3b. DNA methylation is appointed as an epigenetic marker that manage the time and location of genes expression in both normal and diseased cells [33]. In cancers like breast, colon, esophageal, lung, pancreas, ovary, prostate, and other cancers, altered patterns of DNA methylation have been observed [34]. The hypomethylation results in re-expression of silenced genes and genomic instability leading to demethylation of two elements that consists of long interspread transposable elements and short interspread transposable elements [35, 36]. Besides hypomethylation, the outcome of hypermethylation is the silencing of TSGs, such as P15INK4b, P16INK4a, P14ARF, CDH1 or EXT1 [37]
Acetylation and deacetylation
It is well known that the acetylation and deacetylation of N-terminal of lysine residue of histone is a critical part of gene regulation and the process is controlled by two enzymes HAT or HDAC [38]. The acetylation results in condensed chromatin structure leading to cell transcription promotion while deacetylation leads to relaxed chromatin causing suppression of gene transcription [39]. This balance between HAT and HDAC manages the chromatin structure and gene expression [40]. Any imbalance in the activity of HAT and HDAC results in cancer. HAT enzyme is associated with various transcription factors like GCN5-related Nacetyltransferase, MYST, and cAMP response element binding protein (CREB/p300) families. Dysbalances in histone acetylation has been evidenced in Rubinstein–Taybi syndrome, glioblastomas, lung cancers, and AML [41]. On the other side any alteration in expression of different isoforms of HDACs also causes various cancers like increased levels of HDAC 2 and 3 is observed in colon cancer, rise in levels of HDAC 1 is observed in gastric cancer while in lung cancer reduced expression of HDAC5 and HDAC10 is observed [42]. Furthermore, over expression of HDAC 1 is reported in prostate and esophageal squamous cell carcinoma [43].
Histone methylation and demethylation
The extent and location of methylation and demethylation of histones is another important parameter that controls the gene transcription. Both lysine and arginine residues are prone to methylation but lysine residues H3 and H4 of histone tail are more liable to methylation [44]. The known sites for methylation that controls gene activation are H3K4, H3K48 and H3K79 whilst H3K9 and H3K27 are the sites for gene inactivation [45]. A group of proteins containing the SET (enhancer of-zeste, trithorax) called HMT is required by lysine for methylation process [46]. Histone demethylation enzymes known as KDMs are divided into two groups based on their sequence homology and catalytic mechanism. These includes FAD-dependent amine oxidases superfamily called LSDs [47] and (2) the JmjC domain, contains α-ketoglutarate-dependent enzymes, KDMs and Fe(II) [48]. Any irregulatory in epigenetic effects of methyltransferase enzymes can result in a variety of malignancies [49].
Protein phosphorylation
Phosphorylation takes place at side chains of serine, threonine, and tyrosine via phosphate ester linkages in which histidine, lysine and arginine squeeze through the phosphoramidate linkages, and through the mixed anhydride linkages that occur at amino acids, aspartame acid and glutamate [50]. Phosphorylation helps in regulation of a number of biological processes like various signalling pathways, gene expression, cell division, etc. while majority of the cellular functions that includes energy storage, morphological changes, protein synthesis, gene expression, signaling factor release, muscle contraction, and biochemical metabolism are controlled and managed by phosphorylation [51]. A number of signalling pathways are controlled by protein and lipid kinases for regulation of normal cell functions [52,53,54,55,56]. The abnormalities in activity of kinases results in a variety of pathological events, amongst which cancer is the most prominent [52, 53, 56].
Ubiquitination
Ubiquitin system in body consists of three main enzymes ubiquitin-activating enzymes (E1s), binding enzymes (E2s), ligases (E3s), and degrading enzymes [57]. Ubiquitination performs the following functions localization, metabolism, function, regulation and degradation of proteins. The diminished activity of E3 ubiquitin ligase due to some mutations can cause various cancers like renal cell carcinoma, breast cancer, etc. On the other hand, the increase in ubiquitination activity results in cervical cancer. Further total elimination of ubiquitination will lead to colorectal cancer and glioblastoma [58].
SUMOylation
Small ubiquitin-like modified proteins (SUMO) are very similar to ubiquitin proteins as the name signifies. The process of SUMOylation of target proteins results in varied localization and binding partners which ultimately influences the three main parameters: the stability of protein, its transport between cytoplasm and nucleus and regulation of transcription [59]. The promyelocytic leukaemia protein and the oncogenic fusion protein PML–retinoic acid receptor-α are first discovered substrates of SUMO and the occurrence of cancer due to SUMO can be well explained on the basis of the above-mentioned substrates. An infrequent haematological malignancy occurs due to PML-RARα that is called acute promyelocytic leukaemia. The SUMOylation of PML, when distorted, leads to the expression of PML-RARα thus causing APL. SUMOylation is neither tumour promoting nor tumour suppressive rather it is a required process for all cells [60].
Noncoding RNAs in cancer cells
Noncoding RNA is a novel class of genes that control regulatory functions in normal development of cells which get changed in tumor cells. Small nucleolar RNA, PIWI-interacting RNA, small interfering RNA, and microRNA are some of the examples of noncoding RNAs and exhibits functions like transcriptional and posttranscriptional gene silencing via selective base pairing with their targets [4]. Approximately 60% of genes that codes for different proteins and maintains the cellular processes are regulated by miRNAs [61]. Recently, it is reported that miRNAs behave as oncogenes by altering the tumor suppressing proteins or TSGs by modulating the levels proteins that exhibit oncogenic potential [62]. Although, all kinds of ncRNAs exhibits important functions in maintenance of different cellular processes but any irregularity in their function and expression may lead to carcinogenesis [63]. Another ncRNA is small nucleolar RNAs whose dysregulation is reported to be involved in tumorigenesis [64]. For instance, snoRNA42 (H/ACA snoRNA) is a type of snoRNA which is overexpressed in lung cancer [65].
Epigenetics tools for cancer therapy in cancer
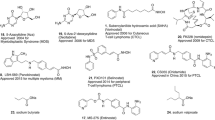
Owing to the well-established role of epigenetic dysregulation towards the origin and progression of cancer, lot of efforts have been invested towards the development of epigenetic drugs for the treatment of cancer. The extensive research conducted on small molecule inhibitors as epigenetic tools (DNMT inhibitors, HDAC inhibitors, DOT1L inhibitors, LSD inhibitors, EZH2 inhibitors, BET inhibitors) makes it evident that the epigenetic proteins are druggable targets. At present, seven agents in three epigenetic target classes (DNMT, HDAC and EZH2 inhibitors) have been approved by the US FDA for the treatment of diverse malignancies (Fig. 1) and a wide range of epigenetic-based drugs are undergoing clinical trials. These include 5-azacytidine (1, DNMT inhibitor approved for the treatment of MDS) [66], 5-Aza-2-deoxycytidine (2, DNMT inhibitor approved for the treatment of MDS) [66], FK-228 (3, HDAC inhibitor approved for the treatment of refractory CTCL) [67], SAHA (4, HDAC inhibitor approved for the treatment of refractory CTCL) [68], PXD101 (5, HDAC inhibitor approved for the treatment of refractory PTCL) [69], LBH589 (6, HDAC inhibitor for the treatment of multiple myeloma) [70] and tazemetostat (7, EZH2 inhibitor approved for the treatment of metastatic or locally advanced epithelioid sarcoma) [71]. Other than the aforementioned FDA approved agents, an anilide type HDAC inhibitor, chidamide (8), has also been approved by CFDA to treat patients with R/R PTCL [72].

FDA and CFDA approved inhibitors of the epigenetic targets
DNMT inhibitors
DNMT blockade is considered to be a successful strategy for the prevention of aberrant DNA hypermethylation. DNMT inhibitors reactivate the aberrantly methylated TSG, thereby causing cancer cells reprogramming that ultimately lead to proliferation arrest and cell death [73, 74]. Literature precedents indicate that various compounds have been identified both at the preclinical as well as clinical level that can erase abnormal methylation patterns via irreversible inhibition of DNMTs, causing proteosomal degradation [75, 76]. This degradation then leads to attenuation of the neoplastic cell phenotype by inducing cell differentiation and tumor cell death [73, 74, 77]. Generally, the inhibitors of DNMT are categorized in to two classes: nucleoside analogs and non-nucleoside analogs (Fig. 2).

DNA methyl transferase inhibitors (nucleoside and non-nucleoside based)
Nucleoside analogs
Comprising of a modified cytosine ring (nitrogen in place of a carbon at 5), nucleoside analogs can be converted to nucleotides and get incorporated into newly synthesized DNA or RNA. The enzyme DNMT gets bound with the analogs through the formation of covalent complexes that leads to the DNA methylation inhibition [78]. 5-Aza cytidine and decitabine (5-aza-2′-deoxycytidine) represents the nucleoside analogues that have been approved by FDA for the treatment of AML and MDS [74, 77]
5-Aza cytidine
5-Aza cytidine (Cytidine analog) is a ribonucleoside analog that undergoes phosphorylation to get incorporated in to the RNA. 5-Azacytidine can also get incorporated into DNA via the ribonucleotide reductase pathway. At present, 5-Aza-cytidine is undergoing several clinical stage investigations for diverse malignancies. A phase 3 clinical trial of azacitidine conducted in patients with higher-risk MDS demonstrated that azacitidine (75 mg/m2 per day, 7 days every 28 days) increased the OS in comparison to conventional care [79]. The phase 4 clinical investigation of azacytidine was also conducted in patients with higher-risk MDS. The study design involved the administration of azacitidine 75 mg/m2/day for 7 days/28-day cycle for up to six cycles. The results of the study demonstrated that out of the 44 patients enrolled for the study, response-evaluable patient (n = 33) did not achieve complete remission or partial remission. However, haematological improvement was attained in 50% patients. RBC transfusion independence was attained in 12 of 32 patients and platelet transfusion independence was achieved in 7 of 18 patients. Neutropenia (52%) and leukopenia (39%) was observed as the common grade 3–4 TAAEs [80] (NCT01201811). In a phase 3 study assessing the benefits of azacitidine over the conventional care regimens in old patients with newly diagnosed AML, it was observed that the treatment with 5-azacytidine (N = 129, 8.9 months) led to remarkable prolongment of the median OS versus conventional care regimens (CCR) (n = 133, 4.9 months) [81] (NCT01074047).
Azacytidine has also been evaluated in various combination regimens. The combination of azacytidine (75 mg/m2) and standard induction therapy was found to be feasible in older patients with AML [82] (Phase 2, NCT00915252). The phase 2 trial of 5-azacytidine with lirilumab (BMS-986015) in patients with refractory/relapsed AML was terminated as the response rate did not meet the anticipated minimum 30%. (NCT02399917). In a phase 2 trial evaluating the combination of 5-azacytidine and sorafenib in older patients (n = 27) with untreated FLT3-ITD Mutated AML, 78% ORR, 26% CR, 44% Cri/CRp and 7% PR was observed. The median OS was 8.3 months and 9.2 months in the 19 responders. Overall, the results demonstrated that the combination was well tolerated in the specified population [83] (NCT02196857). The study evaluating the advantages of sequential azacitidine and lenalidomide in subjects with R/R AML demonstrated that this regimen was only effective in a minority of patients (only 11%). Moreover, significant toxicity was evidenced in some of the cases and three treatment-related deaths occurred [84] (NCT01743859). In an investigation evaluating the efficacy of sequential azacitidine and lenalidomide, or azacitidine in old patients with newly diagnosed AML, it was deduced that the regimen (sequential azacitidine and lenalidomide) is not favoured over azacitidine administered in conventional dose and schedule. With sequential azacitidine and lenalidomide, one-year survival was 44% (95% CI: 28, 60%) where as the one year survival with azacitidine only was 52% (95% CI: 35, 70%) [85] (NCT01358734). In a phase II study conducted in elderly population of higher risk MDS or AML that were, as such, considered unfit for intensive chemotherapy, the combination of azacytidine with escalated doses of lenalidomoide was not well tolerated and was discontinued in majority of the patients owing to toxicity issues. However, some positive results were observed in terms of cytogenetic response in the study. [86] (NCT01088373). In a phase 2 study conducted recently, prophylactic low-dose azacytidine and donor lymphocyte infusions following allogeneic hematopoietic stem cell transplantation for high-risk AML (n = 30 patients) and MDS (n = 10 patients) was evaluated. The study results demonstrated that azacytidine was well tolerated but was discontinued in 20 patients owing to graft-versus-host disease and relapse. The overall and disease-free survivals were 65.5% (CI 95% = 48.2–82.8) at 2 years. On the basis of these results, it was concluded that 5-azacytidine demonstrated potential as a prophylactic treatment to reduce the risk of post-transplantation relapse [87] (NCT01541280). A clinical study for assessing the efficacy of the combination of lirilumab and azacitidine in patients with MDS was conducted and 10 patients were enrolled for the investigation. Two patients achieved CR, 5 achieved marrow CR and 3 demonstrated SD. Grade > 3 AEs (infection or neutropenic fever) were observed in five patients. Overall, the combination of azacitidine and lirilumab demonstrated clinical activity [88] (NCT02599649). Azacitidine in combination with midostaurin in subjects (n = 14 in phase 1 and n = 40 in phase 2, enrolled) with AML and high risk MDS was also evaluated. The study design involved the administration of azacytidine 75 mg/m2 on days 1–7 and midostaurin 25 mg bid (in cohort 1 of phase I) or 50 mg bid (in cohort 2 of Phase I and in Phase II) orally. The results of the study demonstrated that the combination is safe as well as effective for patients with FLT3 mutations that were not previously treated with other FLT3 inhibitors [89] (NCT01202877). In a phase 2 evaluation assessing the combination of azacitidine and etanercept for the treatment of MDS, azacitidine (75 mg/m2/day for 7 days) was administered to twenty-three patients in combination with etanercept (25 mg sc twice a week for 2 weeks every 28 days). The results of the study indicated that a total of 14 patients responded, with CR evidenced in five patients and PR in 8 patients. A hematologic improvement of neutrophils was observed in 1 patient. Overall, the combination was deduced to be endowed with favourable trends in comparison to azacytidine alone. [90]
In a phase 1 study of azacytidine (monotherapy, combination with carboplatin or nab-paclitaxel) conducted in patients with R/R solid tumors, RP2D was determined as 300 mg (every day, days 1–14/21). PR (three/eight) and SD (four/eight) in patients with nasopharyngeal cancer were observed with CC-486 (oral azacitidine) monotherapy. Overall, the study demonstrated that the drug is well tolerated in monotherapy as well as in combination with carboplatin or nab-paclitaxel (NCT02269943) [91]. A phase 3 study was conducted for the assessment of the platelet supportive effects of eltrombopag administered concomitantly with azacitidine. In comparison to azacytidine alone, the combination of eltrombopag and azacitidine led to the worsening of platelet recovery, with lower response rates. Moreover, increased progression to AML was evidenced [92] (NCT02158936). In another recently conducted phase 2 study, the combination of ruxolitinib and azacitidine was found to be safe. Improvement in bone marrow fibrosis coupled with significant spleen response rate was attained in patients with MF. The study design enrolled 46 patients and involved the administration of ruxolitinib twice per day continuously in 28-day cycles for the first 3 cycles followed by the addition of azacitidine (25 to 75 mg/m2, days 1–5) starting with cycle 4 [93] (NCT01787487).
Recently, Onureg (azacitidine 300 mg tablets, CC-486) was approved by US FDA for the continued treatment of adult patients in first remission with AML. The promising results of the AML-001 study (Phase 3 clinical trial) laid the foundation of FDA approval as statistically significant improvement in OS (10 months, median OS time 24.7 months, 95% CI: 18.7–30.5) compared to placebo (median OS time 14.8 months, 95% CI: 11.7–17.6) was attained by the use of onureg. [94] It is noteworthy to mention that a chemical stable analog of 5-Azacytidine, dihydro-5-azacytidine (DHAC), is also biologically active and is relatively less toxic [95, 96].
Decitabine
Decitabine, another nucleoside type DNMT inhibitor, is a desoxyribose analog of cytosine which only gets incorporated in DNA. Decitabine also leads to DNMT depletion and genome hypomethylation. Like, 5-Aza cytidine, decitabine has also been approved by FDA for the treatment of AML and MDS [74, 77]. In a phase II clinical investigation conducted to evaluate the efficacy of decitabine (IV, 15 mg/m2, 5 days–2 weeks) in patients with CML resistant to imatinib mesylate, 35 patients were enrolled (12 in chronic phase, 17 in accelerated phase, and six in blastic phase). The results of the study demonstrated complete hematologic responses in 12 patients (34%) and partial hematologic responses in seven patients (20%). Six patients exhibited major cytogenetic responses, and 10 demonstrated minor cytogenetic responses and the overall cytogenetic response rate observed was 46%. Major adverse effect evidenced was myelosuppression. Overall, it was concluded that decitabine is endowed with clinical activity in imatinib refractory CML [97]. A phase 2 clinical trial for the assessment of decitabine as maintenance therapy for younger adults with AML was conducted in anticipation that 1 year of maintenance therapy would lead to an improvement of disease-free survival for AML patients < 60 years, who as such were not responsive to allogeneic stem cell transplantation in first remission. The results of the study were not encouraging as the maintenance with decitabine did not exert any benefits [98] (NCT00416598). The dynamics of neoplastic cell clearance during decitabine treatment using quantitative monitoring of mutant alleles by pyrosequencing was investigated. The study results demonstrated that the drug was endowed with a noncytotoxic mechanism of action that leads to altered biology of the neoplastic clone and/or normal cells [99] (NCT00067808). A retrospective analysis was conducted to evaluate the response to decitabine in patients with advanced stage MDS. In the study, outcome of patients with baseline marrow blasts ≥ 20% and < 30% (refractory anaemia with Excess Blasts in Transformation—RAEB-t group) and < 20% (MDS group) were compared. A better duration of response was demonstrated by the patients with MDS (9.9 vs. 5 months; P = 0.024) and OS (16.6 vs. 9.0 months) in comparison to patients with RAEB-t [100] (NCT00043381, NCT00260065). A gene expression analysis to assess the gene expression patterns associated with response to decitabine was conducted in a multicenter phase II trial in older AML patients deemed unsuitable for induction chemotherapy. The results of the study indicated that the efficacy of decitabine is partly dependent on immunomodulatory effects [101] (NCT00866073).
In a Phase II study conducted with an aim to assess tosedostat in combination with cytarabine or decitabine in patients (newly diagnosed older) with AML or high‐risk MDS, 34 patients ≥ 60 years old were randomized and tosedostat (120 mg on days 1–21 or 180 mg continuously) was administered with decitabine (20 mg/m2/d) every 35 d. The study outcome indicates that combination of tosedostat and decitabine was tolerated well and resulted in a CR/CRi rate of > 50%. (NCT01567059) [102]. Recently, an inqovi (decitabine and cedazuridine) tablet for treatment of adult patients was approved by US FDA for the treatment of MDS and chronic myelomonocytic leukemia. The approval was attributed to the results of clinical trial that demonstrated similar drug concentrations between intravenous decitabine and inqovi. It was also observed that a considerable proportion of patients that were previously dependent on transfusions did not require the transfusions during an 8-week period. Moreover, intravenous decitabine displayed a similar safety profile to inqovi [103].
Guadecitabine
Guadecitabine, a next-generation hypomethylating agent, is a dinucleotide antimetabolite of a decitabine linked via phosphodiester bond to guanosine. Guadecitabine prolongs the exposure of tumor cells to the active metabolite, decitabine, leading to an enhanced uptake of decitabine into the DNA of rapidly dividing cancer cells. Guadecitabine also offers resistance to degradation by cytidine deaminase [104]. A study (Phase I/II) with an aim to determine the genomic and epigenomic predictors of response to guadecitabine in R/R AML was recently carried out. The study results indicated a 17% response rate to guadecitabine (2 CR, 3 CR with CRi or CR with CRp in the phase I component and 23% (14 CR, 9 CRi/CRp) in phase II. Peripheral blood blasts and haemoglobin were identified as predictors of response and cytogenetics, gene expression, RAS mutations, and haemoglobin as predictors of survival. [NCT01261312, [105]. In a phase 2 study evaluating the combination of guadecitabine with carbotaxol in heavily pretreated patients (n = 100 enrolled) with platinum-resistant recurrent ovarian cancer, promising activity was attained. No serious adverse events were observed in the study. Neutropenia (67%), leukopenia (25%) and anemia (14%) were evidenced as grade 3/4 events. The efficacy evaluation results were as follows: ORR (16%), DCR (37%), PFS (4.1 months), OS (11 months) [106]. In a phase 2 study conducted in patients with HCC, guadecitabine (45 mg/m2) administered on a 28-day cycle was well tolerated in subjects with HCC previously progressed on sorafenib. The study outcome revealed that potent global DNA demethylation (LINE-1) was observed in blood and tumor DNA. To add on, demethylation was seen in patients on promoter of TSG MZB1, which as such, is silenced in HCC [107] (NCT01752933). Recently, the efficacy and safety of guadecitabine was evaluated in phase III study (ASTRAL-1 study) in adults with previously untreated AML. The patients selected were ineligible for intensive induction chemotherapy. The study design involved the administration of guadecitabine, delivered (SC, 60 mg/m2/day for 5 days) in combination with either azacitidine (IV or SC 75 mg/m2/day, 7 days), decitabine (IV 20 mg/m2/day, 5 days) or low dose cytarabine (SC 20 mg bid, 10 days), administered in 28-day cycles. The results of this investigation revealed that primary end points of CR rate or OS were not met (NCT02348489) [108]. Recently, Astex and Otsuka announced the evaluation results of guadecitabine in phase 3 ASTRAL-2 and ASTRAL-3 studies in patients with previously treated AML and MDS or CML. It is disappointing to mention that the guadecitabine did not improve the OS and the study was unable to meet the primary end point [109].
5-Fluoro-2′-deoxycytidine (FdCyd)
5-Fluoro-2′-deoxycytidine represents another deoxyribonucleoside analog that undergoes phosphorylation and is capable of getting incorporated into DNA. The combination of FdCyd and the CD inhibitor tetrahydrouridine (THU) was evaluated in phase I study conducted in cynomolgus monkeys. The results of the investigation indicated that THU administration with FdCyd led to increase in the exposure to FdCyd and improved PO FdCyd bioavailability from < 1 to 24%. Moreover, THU and FdCyd concentrations achieved after PO administration were found to be associated with CD inhibition and hypomethylation, respectively [NCT00378807] [110]. In another phase I investigation of oral 5-fluoro-2′-deoxycytidine with oral THU in patients (N = 40) with advanced solid tumors, FdCyd was administered for 3 − 7 days q wk × 2 in 21-day cycles in combination with THU (administered, PO 30 min prior to Foci). The results of the study are as follows: MTD: FdCyd (160 mg) + THU (3000 mg), 1 × daily days 1 − 6 and 8 − 13, grade 4 toxicities: thrombocytopenia (1 pt), neutropenia (3 pts) and lymphopenia (3 pts), SD: 19 pts [111]. A phase I study was conducted to establish the pharmacokinetic and pharmacodynamics profile of FdCyd (IV) administered with THU (fixed dose − 350 mg/m2) in subjects with advanced cancer. The results of the study are as follows: MTD: Fdcyd (134 mg/m2) + THU (350 mg/m2), days 1–5 and 8–12 every 4 weeks, Phase II dose determined − 100 mg/m2/day FdCyd with 350 mg/m2/day THU, good plasma exposures and the sustained PR was observed at 67 mg/m2/day [112] (NCT00378807). Recently, another study was carried out to evaluate the efficacy of 5-FdCyd in patients with advanced solid tumors. In the study, 93 patients were enrolled (29 breast, 21 head and neck cancer, 25 NSCLC, and 18 urothelial). The outcome of the study was not satisfactory as insufficient responses were achieved and only three PRs were attained. It is noteworthy to mention that the results were only promising in patients with urothelial carcinoma as the preliminary 4-month PFS rate of 42% was attained in the urothelial stratum. In 69% of the patients evaluable for clinical and CTC response, p16-expressing cytokeratin-positive CTCs were increased. Overall, the results observed in this study indicate exploration of FdCyd + THU in future is warranted in urothelial carcinoma [113].
Zebularine
Other than these FDA approved DNMT inhibitors, zebularine (4-Deoxyuridine, ribonucleoside analog), an oral DNA-demethylating drug has demonstrated stability in acidic environments as well as aqueous solutions. Despite being a potential DNMT inhibitor, its clinical translation has been hindered by the limited bioavalability in (< 7%) and primates (< 1%) along with high dose requirements in millimolar concentrations. [77, 114].
Non-nucleoside DNMT inhibitors
Risk of mutagenicity and genomic instability associated with the use of nucleoside DNMT inhibitors [75] has led to the initiation of numerous investigations with an aim of developing nonnucleoside analogs. Most of the non-nucleoside DNMT inhibitors developed so far is small molecule agents that directly target the catalytic sites rather than incorporating into DNA. This section presents a brief account of non-nucleoside inhibitors for natural and synthetic sources.
The sponge Pseudoceratina purpurea yields Psammaplin, a non-nucleoside based dual inhibitor of DNMT and HDAC [115]. A Polyphenol from green tea, EGCG ((-)-epigallocatechin-3-gallate reversibly demethylates methyl-DNA leading to the reactivation of multiple key genes (hMLH1, P16, and RA, in colon, esophageal, and prostate cancer cell lines) [116]. A polyphenolic compound, curcumin, has also been reported to induce global hypomethylation in MV4-11 leukemia cell lines possibly through covalently blocking of the catalytic thiolate of DNMT1, inhibiting DNA methylation [117]. Hydralazine and procainamide have demonstrated tumor suppressor reactivating and antitumor actions in breast cancer [118,119,120]. In a phase II study conducted to combat the issue of chemotherapy resistance in refractory solid tumors, addition of hydralazine and valproate to the same chemotherapy schedule that the patients were receiving, yielded clinical benefits in the selected population. [NCT00404508) [121]. A randomized phase III, epigenetic therapy with hydralazine valproate and chemotherapy in patients with advanced cervical cancer was also carried out. The study design involved the administration of hydralazine (182 mg—rapid acetylators, or 83 mg—slow acetylators along with valproate (30 mg/kg). The study was conducted in 36 patients and four PRs to CT (cisplatin topotecan) + HV (hydralazine valproate) and one in CT + PLA were achieved. SD in five (29%) and six (32%) patients was observed whereas eight (47%) and 12 (63%) showed progression (P = 0.27). Moreover, the study indicated substantial benefits in context of PFS [122] (NCT00532818). Other than these small molecule inhibitors, a second generation phosphorothioate antisense oligodeoxynucleotide, MG98 prevents DNMT1 mRNA translation effects and is under detailed preclinical studies and clinical stage investigations (phase I/II clinical trials) in solid tumors [123,124,125]. Another, small molecule inhibitor, RG-108 is reported to directly inhibit DNMT1 catalytic domain and block DNMTs without causing enzyme degradation [115, 126]. Disulfiram was also identified as a DNMTi as it was found to reduce global 5mC levels, as well as demethylate and reactivate the expression of epigenetically silenced TSGs [127]. SGI-1027, a quinolone based compound, exhibited inhibitory potential towards DNMT1, DNMT3A and DNMT3B, leading to demethylation and reactivation of TSGs [128]. Table 1 presents the clinical update of DNMT inhibitors undergoing clinical stage investigations.
EZH2 inhibitors
EZH2, a crux subunit of the PRC2, is a HMT enzyme responsible for methylating lysine 27 (mono-, di- and trimethylation) in histone H3 (H3K27). H3K27me3 is more frequently interlinked with transcriptional repression, and it is a significant epigenetic phenomenon during tissue development and stem cell fate determination. Specifically, functioning of EZH2 in biological processes occurs through 3 types of mechanism viz. PRC2-dependent H3K27 methylation, PRC2-dependent non-histone protein methylation, and PRC2-independent gene transactivation [129,130,131,132,133,134,135,136]. As such, EZH2 works as a master regulator of cell cycle progression [137], autophagy, and apoptosis [138], promotes DNA damage repair and inhibits cellular senescence [139]. In view of the aforementioned notions, it is evident that EZH2 plays an important role in cell lineage determination and relative signalling pathways. The enzyme has been found to be overexpressed in wide varieties of cancer, such as prostate, liver, gastric, breast, bladder, lung, and pancreatic cancers [129,130,131,132,133,134,135] with literature precedents ascertaining the role of EZH2 in augmenting the development and progression of cancer. In this context, EZH2 targeting therapies, at present, have garnered significant attention for the treatment of many types of cancer. Till now, tazemetostat stands as the only approved EZH2 inhibitor for advanced epithelioid sarcoma, however, various EZH2 inhibitors are being evaluated at preclinical and clinical stages. The details of selected important EZH2 inhibitors are mentioned in Table 2 and the structures are shown in Fig. 3. It is noteworthy to mention that a homolog of EZH2, EZH1, is present in a non-canonical PRC2 complex. As such, EZH1 complements EZH2 in mediating H3K27 methylation and is also endowed with HMT activity. In light of this disclosure, it is highly anticipated that simultaneous inhibition of EZH1/EZH2 that can be attained via dual EZH1/EZH2 inhibitors might exert potent anticancer effects. A brief mention of a dual EZH1/EZH2 inhibitor is also included in Table 2 [140].

EZH2 inhibitors
The results covered in this Table 2 indicates that EZH2, at present, is considered as potential target for the design of cancer therapeutics and many EZH2 inhibitors are under development and evaluation in clinical trials. Other than the promising results of EZH2 inhibitors in monotherapy, combination of EZH2 inhibitors with immunotherapy or chemotherapy has also demonstrated synergism and is likely to be the futuristic strategy to extract therapeutic benefits for EZH2 inhibition. Researchers are also employing rational approaches for accomplishing new EZH2 inhibitors that can display high efficacy and low selectivity.
DOTIL inhibitors
H3K79 (Methylation of histone 3 at lysine 79) (H3K79) is one of the main mechanisms involved in gene expression. HMT DOT1L targets the histone H3lysine 79 (H3K79) residue for mono-, di- and tri-methylation. As such, DOT1L has a critical role in the regulation of gene transcription, development, cell cycle progression and DNA damage repair. Specifically, DOT1L leads to enhanced H3K79 methylation, methylation of open chromatin, downstream oncogenes overexpression and leukemogenesis via interaction with mixed lineage leukemia [165]. Studies have revealed that changes in normal expression levels of DOT1L have been found in prostate, breast, and ovarian cancer. In addition, H3K79me levels are elevated in AML patients bearing MLL rearrangements [166]. In light of the aforementioned, attention has been paid towards the development of small molecule DOT1L inhibitors and accordingly pinometostat (Fig. 4), a potent and selective small molecule DOT1L inhibitor endowed with subnanomolar affinity for DOT1L and > 37 000-fold selectivity against towards HMT has been investigated at the clinical stage [167,168,169,170]. The results of the preclinical studies indicated that it selectively inhibits intracellular H3K79 methylation in a concentration- and time-dependent manner. Pinometostat demonstrated activity against leukemia involving MLL-r in in vivo rodent xenograft studies [171,172,173]. Subsequently, a phase 1 study of pinometostat (dose escalation study) was performed in subjects with R/R MLL-r leukemia. The study design involved the administration of pinometostat via continuous intravenous infusion until disease progression or unacceptable toxicity. A total of 18 patients were enrolled for the investigation, 9 of them received 70 mg/m2/day of pinometostat, 7 patients were dosed at 90 mg/m2/day and the 2 patients were dosed at 45 mg/m2/day. The results of the study revealed that DLTs were observed which included hypocalcemia; hypophosphatemia; apnea, elevated transaminase, drug related AEs: anemia; thrombocytopenia; neutropenia; leukopenia; rash; lymphopenia; ALT elevation; nausea; vomiting. The drug demonstrated an acceptable safety profile and RP2D was determined as 70 mg/m2 CIV in children > 1 yr. As such, no objective responses were observed. (NCT02141828) [174]. In another phase 1 study, evaluating the efficacy of pinometostat (EPZ-5676) administered as continuous intravenous infusion in patients with MLL-r leukemia (adults). 51 patients were enrolled and CR was observed in 2 patients. Nausea, constipation, febrile neutropenia (grade 1 & 2) were observed as AEs. Overall, the results demonstrated that pinometostat was safe and endowed with modest efficacy in monotherapy. MTD was not determined in the study (NCT01684150). [175]

Pinometostat
Other than the completed studies mentioned above, pinometostat is also undergoing clinical evaluation in combination with azacytidine in subjects with R/R, or newly diagnosed AML leukemia with 11q23 rearrangement (Phase 1/2 Studies, NCT03701295, status recruiting) and a Phase 1b/2 investigation in combination with standard induction chemotherapy in patients with newly diagnosed AML with MLL rearrangement (NCT03724084, status – recruiting).
HDAC inhibitors
It is well known that post translational modifications of histones are involved in cancer development and progression via modulation of gene transcription, chromatin remodeling and nuclear architecture. Tightly controlled by the opposing activities of HATs and HDACs, histone acetylation is a well explored post translational modification. As such, HDACs removes the acetyl groups on N-terminal lysines of the histone proteins, thereby inducing histone hypoacetylation that leads to loosening of the chromatin structure ultimately facilitating the transcriptional process [176,177,178,179,180,181,182,183,184,185,186].
Eighteen HDACs in humans are categorized in to two families based on their catalytic mechanisms. Out of the 18, 11 are zinc dependent metalloenzymes (HDAC1-11) that catalyse the hydrolysis of amide bond using water as a nucleophile while the other 7 are sirtuins (1–7) that employ NAD + as a cofactor and transfer the acyl group to the ribose sugar (C2 position). The 18 human HDACs are further delved in to four classes. Class I (HDAC1, HDAC2, HDAC3 and HDAC8) Class IIa (HDAC4, HDAC5, HDAC7 and HDAC9) Class IIb (HDAC6 and HDAC10) Class III (sirtuins 1–7); Class IV (HDAC11) [176, 187,188,189]. Numerous studies have revealed that class I, II and IV are aberrantly expressed in diverse malignancies that majorly include solid and hematological tumors. Owing to this, HDACs as drug targets in cancer have been exhaustively explored and HDAC inhibitors are considered to the key for epigenetic cancer therapy owing to their ability to induce relief of transcriptional repression in various leukemias [177].
Structural fabrication of HDAC inhibitors includes a cap group, linker part and a Zn-binding group and HDAC inhibitors are generally categorized in to two structural classes on the basis of the zinc binding group: Hydroxamic acids and the aminoanilides. SAHA [68], Belinostat (PXD101) and Panobinostat (LBH589) [70] represent the US FDA approved hydroxamic acid type HDAC inhibitors and mocetinosat [190], MS-275 [191] and chidamide [72] are the representative examples of N-(2-Aminophenyl)benzamide binding unit containing HDAC inhibitors.
In 2006, suberoylanilide hydroxamic acid (SAHA) became the first HDAC inhibitor to get FDA approval for the treatment of CTCL. Spurred by the success of SAHA, several HDAC inhibitors with linear methylene chains have progressed to clinical stage investigation viz tefinostat [192], CG200745 [193], ricolinostat [194], citarinostat [195], CUDC-101 [196] and tinostamustine [197]. Another HDAC inhibitor, romidepsin [67] belonging to the family of depsipeptide natural products was approved for CTCL. Sipruchostatin, also belonging to the class of natural product depsipeptide is currently undergoing phase 1 clinical investigation for the treatment of solid tumors [198, 199]. Belinostat (PXD-101) that bears a more rigid alkenyl hydroxamic acid is the third HDAC inhibitor to be approved by US FDA in 2014 [69] for the treatment of PTCL. Another alkenyl type small molecule HDAC inhibitor, Panobinostat, received FDA approval in 2015 for the treatment of patients with multiple myeloma [70]. The FDA approvals of belinostat and panobinostat opened an avenue for the exploration of alkenyl type drug candidates and subsequently, resminostat [199,200,201,202] and pracinostat [203,204,205] were identified that are now undergoing clinical stage investigation. Givinostat [206,207,208], abexinostat [209, 210], AR-42 [211, 212] and bisthianostat [213, 214] represents the chemically related compounds bearing a more rigid phenylhydroxamic acid while quisinostat [215,216,217], nanatinostat [218, 219] and fimepinostat [220] containing a more polar pyrimidinyl heteroaromatic hydroxamic acid have also demonstrated substantial efficacy.
Ortho-aminoanilides constitute the other class of synthetic HDAC inhibitors that exhibit bidentate coordination of the carbonyl oxygen and aniline nitrogen with the active site zinc cation. Ortho-amino anilides are weak metal binders than hydroxamic acids and exhibit unusual kinetics along with slow and tight binding to HDACs. Tacedinaline [221], etinostat [222, 223], mocetinostat [224, 225], tucidinostat [226, 227], domatinostat [228] and CXD1 [229] represents the clinical candidates from this class of synthetic HDAC inhibitors. Among the benzamides, only tucidinostat (chidamide) stands as the approved benzamide (CFDA approved) for the treatment of patients with recurrent or refractory PTCL [226].
Some of the HDAC inhibitors such as CKD-504, 506, CS3003, HG116, KA2507, OK-179 (undisclosed structures) are also undergoing phase I clinical investigation. All of them belongs to the category of selective HDAC6 inhibitors except OKI-179 which is a synthetic analogue of largazole, isolated from a marine cyanobacterium of the genus Symploca, (selective Class I HDAC inhibitor) [224].
A clinical/preclinical update of the HDAC inhibitors is presented in Table 3 and the structure of the HDAC inhibitors are shown in Fig. 5.

HDAC inhibitors
The results presented in Table 3 clearly indicate that HDAC inhibitors have demonstrated promise in oncology indications as single agents. Selective toxicity in liver cancer exerted by tefinostat [236], encouraging results of clinical trials evaluating ricolinostat [230] in multiple myeloma, motivating results attained with aromatase inhibitor, exemestane and the orthoaminoanilide entinostat in the treatment of postmenopausal breast cancer [237], combination of exemestane and tucidinostat receiving NMPA approval for the treatment of breast cancer [227], advancement of the combination of pracinostat and azacytidine to higher stage clinical investigation for the treatment of MDS and newly diagnosed AML [238, 239] exemplify some of the successful clinical stage investigations trials conducted in the recent past.
LSD1 inhibitors
Histone demethylase LSD 1 is reported to be overexpressed in diverse malignancies. Growth inhibition of multiple tumor types is exerted via inactivation or knockdown of LSD1 in cancer cells [240,241,242,243]. Literature survey indicates that only those inhibitors that specifically targets lysine specific histone demethylase 1A (KDM1A) have been able to advance to clinical stage investigation [244, 245]. In light of the aforementioned, inhibition of LSD1/KDM1A is presently being given serious consideration for the fabrication of new antitumor scaffolds. The pipeline of LSD1 inhibitors is filled with numerous candidates undergoing clinical stage investigation in monotherapy as well as combination therapy namely tranylcypromine, phenelizine sulfate, ORY-1001, GSK-2879552, IMG-7289, INCB059872, CC-90011, and ORY-2001 (Fig. 6). This section will present an overview of the results of clinical trials of the LSD1 inhibitors (Table 4).

LSD1 inhibitors
BET inhibitors
The bromodomain and extra-terminal domain (BET) family of bromodomain containing proteins are epigenetic readers that are considered to be important regulators of the epigenome owing to their ability to recognize N-acetyl lysine (KAc) post translational modifications on histone tails [269]. Among the bromodomain containing proteins, BET family (BRD2, BRD3, BRD4, and BRDT) has been the most extensively explored and the results of these explorations has ascertained its links with diverse cancers. In light of the aforementioned, the BET family of proteins represents a well-established therapeutic target for oncology and immunoinflammation indications [270,271,272] and numerous small molecule inhibitors capable of abrogating the BET − KAc interactions are currently under clinical investigation (Fig. 7). This section will present an update on BET inhibitors undergoing clinical studies.

BET inhibitors
JQ1 is a thienotriazolodiazepine that represents the first generation synthetic BET inhibitor. It potently inhibits BRD2, BRD3, BRD4, and the testis-specific protein BRDT in mammals. JQ-1 is endowed with anticancer efficacy in murine and xenograft models of NMC, AML, multiple myeloma, and Burkitt's lymphoma. The preclinical study results have reported toxicities associated with the use of JQ1 that included intestinal crypts disruption in mice with reduced BRD4 levels, impaired long-term memory and heightened anxiety, depletion of hematopoietic cells, skin hyperplasia, and; neuronal defects and obesity in mice with reduced BRD2 levels. Though JQ1 is not being evaluated at the clinical level, several structurally related BET inhibitors are being clinically evaluated for diverse malignancies [273].
OTX-015 is a thienotriazolodiazepine BET inhibitor that selectively blocks BRD2/3/4 and is the first BET inhibitor to be evaluated clinically [274]. It is endowed with striking antitumor activity against a panel of cell lines derived from hematologic malignancies and solid tumors (breast and prostate cancer, neuroblastoma and glioblastoma [275,276,277,278,279,280,281]. OTX015 was administered orally in a dose-escalation, phase Ib study in patients with haematologic malignancies. In the study, analysis of the blood samples from 81 patients was performed that were administered OTX015 (dose: 10–160 mg or 40 mg twice daily). The results of the study demonstrated that OTX015 pharmacokinetics can be adequately described by a one compartment open model with linear elimination. The absorption rate constant (ka) = 0.731 h-1, V = 71.4 L and CL = 8.47 L h-1 were the estimated pharmacokinetic parameters. Overall, the results from population pharmacokinetic modelling of OTX015 plasma concentrations indicated that there is need for dose adjustment [282] (NCT01713582). In a dose escalation study conducted in patients with acute leukaemia, OTX015 was administered orally at doses increasing from 10 mg/day to 160 mg/day (14 of 21 days). The results of the study recommended the administration of 80 mg OTX-015 (once daily) on a 14 days on, 7 days off schedule [283, 284] (NCT01713582). OTX-015 demonstrated a dose-proportional exposure and a favorable tolerance profile in solid tumor patients along with reasonable activity in NUT midline carcinoma patients (Phase 1b clinical trial). In addition, clinical improvement was evidenced in heavily pre-treated castrate-resistant prostate cancer patients. The RP2D was deduced to be 80 mg QD [oral OTX-015 continuously (21/21)] [285] (NCT02259114). A Phase 1 study of OTX-015/MK-8628 in patients with advanced solid tumors (NCT02698176) and a phase 2 study of OTX-015/MK-8628 in GBM patients were terminated owing to lack of clinical activity/ limited efficacy and some safety related concerns (NCT02296476). Another phase 1 clinical study of OTX015/MK-8628 in patients with selected hematologic malignancies (MK-8628-005) is currently active (not recruiting) (NCT02698189).
CP-0610 CP-0610 is a potent and selective benzoisoxazoloazepine BET bromodomain inhibitor. A preclinical study results demonstrated that CP-0610 attenuated BET-dependent gene expression in vivo and was found to be endowed with antitumor efficacy in an MV-4-11 mouse xenograft model [286]. A first-in-human Phase 1 study of CPI-0610, was conducted in subjects with R/R lymphoma. The study design involved oral administration of CPI-0610 once daily (QD) on days 1–14 of a 21-day cycle. The number of patients enrolled were 44. The results of the study demonstrated that CPI-0610 was well tolerated. However, reversible and non-cumulative dose dependent thrombocytopenia was the principal toxicity evidenced. Though the study results could not determine MTD, clinical efficacy was observed [287]. A phase 1 study (study design: 3 + 3) of CPI-0610 (dose = 6–300 mg) in capsule form and at 125 mg or 225 mg in tablet form in patients (n = 64) with progressive lymphoma was conducted. Determination of MTD was the primary end point of the study which was deduced to be 225 mg in tablet form. Thrombocytopenia (42.2%), nausea (17.2%), and fatigue (17.2%) were the common TAAEs. The efficacy analysis results conducted in 38 patients demonstrated 5 OR (13.2%), including 2 CR and 3 PR. (recruitment status: complete, NCT01949883) [288]. CPI-0610 was also evaluated clinically in a phase 1 study (study type interventional) in subjects (30 participants) with previously treated multiple myeloma. The study was initiated on July, 2014 and completed on November 2017 (study results—not posted yet, NCT02157636). A phase 2 clinical investigation of CP-0610 in patients with malignant peripheral nerve sheath tumors was recently withdrawn due to lack of enrolment (NCT02986919). In light of the significant clinical activity evidenced preclinically and clinically (Phase 1 studies), CP1-0610 is currently being evaluated in monotherapy and in combination with ruxolitinib in patients with MF (NCT02158858). Evaluation of the spleen volume response and effects on transfusion independence rate are the primary objectives of the study. [289].
BMS-986158 a BET bromodomain inhibitor that downregulates c-Myc expression and causes cancer cell death in c-Myc-driven cancer cell lines in vitro. It is endowed with inhibitory potential towards tumor growth in triple-negative breast, colorectal, and lung cancer patient-derived mouse xenograft models [290]. In a phase I/IIa dose escalation study (advanced cancer), BMS-986158 (0.75, 1.25, 2.0, 3.0, or 4.5 mg) was administered to patients (n = 69, once daily). BMS-986158 displayed a Tmax of 2–4 h, T1/2 of 33–82 h, and linear PK. Reversible thrombocytopenia was observed as the DLT. Adverse events were observed in 63% patients (diarrhea—34%, thromobocytopenia, 28%, fatigue 16%). Out of the four patients with NMC, (dose: 2 mg, Schedule A, 279 days), one patient experienced a 16% tumor reduction. CCR2 and HEXIM1 were the biomarkers examined [291]. BMS-986158 is currently undergoing evaluation in pediatric cancer (NCT03936465, status recruiting) and a phase 1/2 clinical evaluation to assess safety, tolerability, pharmacokinetics, and pharmacodynamics of BMS-986158 (advanced cancers) (NCT02419417, status recruiting).
ZEN003694
It is an orally available small molecule selective BET bromodomain inhibitor ZEN-3694 that demonstrates > 20-fold selectivity over non-BET bromodomains, thereby causing an inhibition of interaction of acetylated histone peptide at IC50 values in low nanomolar range). ZEN-3694 displays synergistic efficacy with many standard of care and targeted therapies in diverse malignancies and is particularly effective against CRPC and TNBC xenograft models [292]. The safety and tolerability of ZEN003694 (phase 1 study) in patients (n = 44) with mCRPC (NCT02705469) was completed on October 2017 (results not posted yet).
The combination studies of ZEN-3694 and enzalutamide in patients with mCRPC was conducted. The study design involved a 3 plus 3 dose escalation scheme (starting dose: 36 mg ZEN-3694 + ENZ 160 mg, daily oral dose). Doses were expanded in parallel cohorts (ZEN-3694 −48 and 96 mg daily) and 64 patients were enrolled in the study. Overall, the outcome of the study was quite optimistic and demonstrated that ZEN-3694 is endowed with an acceptable safety and PK profile. The combination could also attain promising disease stabilization warranting its further investigation. The study results are summarized as: adverse events—transient photophobia (66%), nausea (40%), fatigue (31%), decreased appetite (22%), and dysgeusia (16%). No Grade ≥ 3 thrombocytopenia was observed, overall median TTP was determined to be 44.4 weeks (NCT02711956) [293]. A phase 2 study of ZEN-3694 in combination with talazoparib in patients with TNBC without germline mutations of BRCA1 or BRCA2 is currently recruiting (NCT03901469). The clinical evaluation of ZEN-3694 in combination with enzalutamide plus pembrolizumab in patients with mCRPC is expected to start on August 3rd, 2020 (Current status: not yet recruiting). The study design is as follows: clinical stage: Phase 2, Study type: open lable, non randomized Interventional, Enrollment (estimated): 54.
Molibresib (GSK525762)
Endowed with antiproliferative effects evidenced in preclinical studies models of NC and other solid and hematologic malignancies, GSK25762 is an orally bioavailable, small-molecule BET inhibitor [294]. The phase 1 study of GSK525762 was conducted in patients with nuclear protein in testis (NUT) carcinoma (NC) and other solid tumours to determine the RP2D and establish the pharmacokinetic as well pharmacodynamics profile. The study design involved administration of GSK525762 (orally once daily, 3 + 3 design, dose escalation study, n = 65) starting with a dose of 2 mg/d. Grade 4 thrombocytopenia encountered with molibresib 60–100 mg along with gastrointenstinal events were the most frequent TAAEs. 80 mg once daily was determined as RP2D. GSK52576 exhibited rapid absorption and elimination (maximum plasma concentration: 2 h; t1/2: 3–7 h). Reductions in circulating monocyte in circulating monocyte chemoattractant protein-1 levels were also observed. Four patients out of 19 patients attained confirmed or unconfirmed PR, eight had SD stable and four were progression-free (> 6 months) [295] (NCT01587703). Clinical activity was also evidenced in a phase 1 trial conducted in patients (n = 46) with AML. The results of the study are as follows: PRs—3, CRs—2, AEs—dysgeusia (37%), diarrhea (33%), and nausea (28%). Overall the drug was found to be safe and effective. [296] An open label study in patients with NHL was conducted and the RP2D was determined as 60 mg QD. Response rate (50%) was observed in patients with CTCL. Manageable thrombocytopenia was observed that was monitorable and reversible. [297]. Evaluation (pharmacokinetics, pharmacodynamics and clinical activity) of GSK525762 (dose escalation study) for R/R hematologic malignancies has recently been completed, however, the results have not been posted yet. (NCT01943851). GSK525762 is currently being evaluated in combination with fulvestrant in patients with HR + / HER2− advanced or metastatic breast cancer. The expected completion date of the study is September 28th, 2020 (NCT02964507, status: active, not recruiting). GSK525762 is also currently being evaluated in combination with androgen deprivation therapy and other agents in patients with castrate-resistant prostate cancer (NCT03150056, status- active, not recruiting).
GSK2820151
A potent and selective small molecule BET inhibitor, GSK2820151, has demonstrated inhibitory potential towards the proliferation of several solid tumour cell types along with in-vivo activity in xenograft models [298]. A phase 1 clinical investigation of GSK2820151 in patients with advanced or recurrent solid tumours was recently terminated to focus on the clinical growth of another BET inhibitor, GSK525762, owing to established risk benefit profile. (NCT02630251).
INCB054329
A structurally distinct BET inhibitor that has demonstrated potency against B-cell malignancies in preclinical models. A phase 1/2 study of INCB054329 in patients with advanced malignancies was terminated due to pharmacokinetic issues (NCT02431260).
INCB057643
INCB057643 is a potent and selective small-molecule BET inhibitor [299]. Phase I/II clinical trial was conducted in patients with solid tumor (NCT02711137) and the study involved the oral (continuous) administration of INCB057643 (once daily) in 21 days cycles (3 + 3 design). The results of the study are as follows: TAEs—increased conjugated bilirubin, increased INR, TCP, decreased appetite and dyspnea, 1 patient with SD ≥ 6 months (11 of 13 patients evaluable for efficacy in part 1). 2 patients (evaluable for efficacy, Part 2) of 14 patients with solid tumors had PD. Overall, the outcome of the study demonstrated clinical activity of INCB057643. The study is now terminated owing to safety issues [NCT02711137)] [300]. Clinical investigation of INCB057643 in patients with MF started on August 31, 2020. (NCT04279847). A phase 1/2 trial was started on January 26 2017 to evaluate the efficacy of pembrolizumab, epacadostat with INCB057643 (NCT02959437).
ODM-207
ODM-207 is a novel, potent and highly selective BET bromodomain inhibitor. In preclinical studies, it has demonstrated significant efficacy against prostate and breast cancer. [301]. ODM-207 was evaluated for safety and pharmacokinetics in patients with selected advanced solid tumours (Phase 1/2 study) (NCT03035591). The study has been completed but the results have not been published yet.
RO6870810
RO6870810 is a small-molecule BET inhibitor. A clinical stage investigation (Phase 1, two parts) was conducted to evaluate the safety, PK, tolerability, and efficacy in subjects (n = 84, 54 in part A and 30 in part B) with advanced solid tumors. The study was completed on October 2017, but results have not yet been published (NCT01987362). Another phase 1 study of RO6870810 in subjects with AML and MDS was completed on August 2017 but the results have not been posted. (NCT02308761). The phase 1 studies of RO6870810 (mono and combination therapy in advanced multiple myeloma was also recently completed. (NCT03068351). A combination study of RO6870810 and Atezolizumab (PD-L1 Antibody) in participants with advanced ovarian cancer or TNBC was terminated (NCT03292172). A combination study (phase 1) of RO6870810 and venetoclax, with or without Rituximab, in patients with DLBCL was recently completed but the results have not been posted. (NCT03255096).
BAY 1238097
BAY 1238097 is a highly selective and potent BET inhibitor. In a preclinical study, BAY 1238097 demonstrated tumour growth inhibitory potential in xenograft mouse models of lymphoma [35]. A phase 1 study of BAY1238097 was conducted in patients with cytologically or histologically confirmed advanced refractory malignancies. BAY 1238097 was administered orally (twice weekly in 21-day cycles) employing a dose escalation strategy with a starting dose of 10 mg/week. The results of the study demonstrated prolonged SD in two patients (no responses observed). Increased HEXIM1 expression and decreased MYC expression was also observed. The study was terminated owing to the occurrence of DLTs at dose below targeted drug exposure. (NCT02369029) [302].
CC-90010
It is an oral and reversible BET inhibitor endowed with significant activity in lymphoma and solid tumor cell lines [303]. CC-90010 was evaluated in subjects with advanced solid tumors and R/R NHL (R/R NHL). In the study, 69 patients were enrolled (67 with solid tumors and 2 with R/R NHL). In 17 patients, grade 3/4 TAEs were observed that included (≥ 2 pts) thrombocytopenia (7%), platelet count decreased (4%), fatigue (3%), and increased alanine aminotransferase (3%). PR was observed in 2 patients; prolonged SD was observed in 7 patients (SD > 9 months). Overall, preliminary clinical activity was observed in the study with manageable TAAEs. (NCT03220347) [303]. The phase 1 clinical evaluation of CC-90010 in progressive/recurrent diffuse astrocytoma, anaplastic astrocytoma and glioblastoma started on August 2, 2019 (Current status, NCT04047303). Phase 1 combination studies of CC-90010 with temozolomide (with or without radiation therapy) in patients with newly diagnosed glioblastoma is underway (current status—not yet recruiting, NCT04324840). A phase 1/2 investigation of CC-90011 in combination with cisplatin and etoposide in patients with SCLC is also underway (status—recruiting, NCT03850067).
AZD5153
A novel, reversible BRD4 inhibitor endowed with bivalent mechanism of action and substantial antitumor potential as evidenced in preclinical studies. In a phase 1 dose escalation study, oral AZD5153 QD/BID was administered to patients with RR solid tumor, including lymphoma. 28 patients were treated in 7 cohorts. The results of the study demonstrated that treatment related AEs were observed in 50% patients. Moreover, a linear increase in PK was evidenced. Overall, it was concluded that AZD5153 as a single agent is safe and well tolerated (30 mg QD and 15 mg BID) (NCT03205176) [304] AZD5153 is also undergoing phase 1 studies in combination with Acalabrutinib in patients with R/R aggressive NHL (NCT03527147).
FT-1101
A promising pan-BET inhibitors possessing equipotent inhibition for BRD2, BRD3, BRD4, and BRDT. FT-1101 exerts substantial anti-proliferative effects against a panel of human leukemia cell lines. FT-1101 has demonstrated higher tumor growth inhibition (TGI) in xenograft and syngeneic models in a relative comparison with JQ1 [305, 306]. Phase 1 clinical trial of FT-1101 (monotherapy and in combination with Azacitidine) in patients with R/R hematologic malignancies got completed in March 2019. [NCT02543879]. The study involved the administration of oral FT-1101 (10–600 mg) dosed once a week, every other week or monthly during dose escalation. The results of the study indicated that 1 patient (out of the 30 evaluable patients) on the 400 mg (every other week) schedule showed complete remission with incomplete hematologic recovery and 19 pts attained SD. Only one patient among the evaluable NHL patients (n = 3) achieved SD. Overall, FT-1101 was found to be safe and exhibited acceptable PK and modest clinical activity in R/R AML/MDS and NHL pts [306] (NCT0254387).
PLX51107
An orally active small molecule inhibitor that blocks interactions mediated by the four BET family proteins at low nanomolar potency [41]. A study of PLX51107 (3 + 3 dose escalation study, phase I) was conducted in subjects with R/R solid tumors (lymphomas included) and AML. The study design and results are as follows: study type: 3 + 3 dose escalation study (Phase 1) type of administration: oral, continuous (QD and BID), number of patient enrolled = 36, common AEs (in ≥ 15 pts)—fatigue (33%), vomiting (25%), diarrhea (25%), nausea (19%), bilirubin increase (17%) and INR increase (17%); Results: 8 patients out of 36 pts achieved SD (2 uveal melanoma, 3 sarcomas, 1 CRPC, 1 NSCLC) ranging 4–14 months [307] ((NCT02683395). The study has been terminated owing to business related reasons. The phase 1 combination studies (PLX51107 and Azacitidine) for the treatment of patients with AML or MDS to determine the minimum safe and biologically-effective dose was started on September 2019. (Status—recruiting, NCT04022785).
SF1126
SF-1126 is a dual inhibitor of phosphatidylinositol-3-kinase and BRD4. It exerts simultaneous disruption of two key MYC-mediating factors that promote cancer cell growth (NCT03059147). SF1126 was administered IV days 1 and 4, weekly in 28 day-cycles in phase I study in subjects (n = 44) with advanced solid tumours and B-cell malignancies. The results of the study are as follows: toxicity: grade 1 and 2 toxicities with a single DLT at 180 mg/m2 (diarrhea); best response: stable disease in 19 of 33 (58%) evaluable patients; maximum administered dose (1110 mg/m2). Overall, SF1126 was well tolerated [308]. A phase 1 study of SF1126 with R/R neuroblastoma was terminated due to low patient accrual (NCT02337309). The clinical evaluation of SF1126 in combination with nivolumab in patients with advanced HCC was initiated on March 27, 2017 (status—active not recruiting, NCT03059147). A phase II Study SF1126, in patients with recurrent or progressive SCCHN and mutations in PIK3CA gene and/or PI-3 kinase pathway genes was terminated due to slow enrolment (NCT02644122).
Medicinal chemist’s perspective: prudent approaches to steer the progress of the epigenetic inhibitors from preclinical to clinical level
The clinical advancement of numerous epigenetic inhibitors as discussed in the preceding sections coupled with the better understanding and growing insights in context of the role of epigenetic mutations in cancer clearly renders ample scope for the medicinal chemist to expand the size of the armoury (epigenetic inhibitors) at the preclinical level. On the precedential basis, it has been well evidenced that only a few preclinical active scaffolds are able to replicate their promising activity profile at the clinical level. Thus it becomes imperative to have a sagacious understanding of drug design at the root level for rationally constructing epigenetic inhibitors employing diverse strategies. Much to the delight of the medicinal chemist, most of the epigenetic targets are druggable and small molecules have been exploited exhaustively to bind to these targets in pursuit of attaining therapeutic benefits in cancer. In this section, we discuss the potential structural engineering approaches citing several selected literature precedents that appears to be promising to push the preclinical pipeline of the epigenetic drugs to the clinical level by leaps and bounds. The furnishment of new inhibitors is most likely to be achieved through the below mentioned pragmatic approaches:
Proteolysis-targeting chimera “PROTAC”
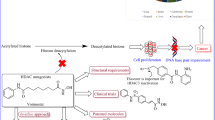
Growing inclination towards the development of enzyme/protein degraders appears to be most pragmatic way to carry forward the growth of epigenetic therapy. Literature precedents reveals that the degradation of the target protein can yield enhanced therapeutic benefits at low concentrations and thus futuristic attempts should be majorly inclined towards the development of epigenetic inhibitor based PROTACs. As such, PROTACs are hetero bifunctional small molecules composed of three chemical elements namely ligand for the target protein, ligand for E3 ubiquitin ligase and a linker for the tetheration of the two ligands (Fig. 8) [309]. Engineered to induce degradation of disease causing proteins by ubiquitin proteasome pathway, PROTAC degrade disease-causing proteins through the cell’s ubiquitin/proteasome system and functions by recruiting an E3 ligase to tag the target protein for ubiquitination. Gratifyingly, PROTACs can induce degradation of the target proteins at low exposures and are catalytic in their mode of action [309,310,311].

PROTAC design
Epigenetic drug discovery field, at present, is flooded with numerous agents that have shown promise at preclinical or early stage clinical level. An obvious step forward to extract the benefits of these inhibitors is their installation in the PROTAC model as ligands for the target protein (Fig. 8). Most classes of epigenetic inhibitors consist of simple chemical architectures that offer ample scope to tether them to the ligand for the E3 ligase through a linker. Moreover, epigenetic targets comprise of a wide range of enzymes (HDAC, LSD1, DOT1L, EZH2, BET proteins, DNMT) and this fact can be leveraged for the design of libraries of PROTAC based scaffolds that can demonstrate amplified antiproliferative effects. Practically, quest to accomplish epigenetic inhibitors based PROTACS will require a selection of a target followed by recruitment of a structure (ligand for the target protein) that not just is efficacious in terms of binding to an enzyme/protein or receptor but also bears a suitable site for tetheration with the E3 ligase ligand via a linker. The recruitment of ligand for the E3 ligases is a difficult task as there are limited options (Fig. 9) available in context of the chemical architectures. Usually, the expression pattern of E3 ligases in a particular tissue (Table 5) is observed to select the ligand for E3 ligase. For instance, high expression of E3 ligase CRBN is reported in lung cancer, thus ligands for CRBN (lenidomide, thalidomide, pomalidomide) should be installed in the PROTAC model to develop an anti-lung cancer therapeutic. Likewise, VHL ligand requires to be accommodated in the PROTAC template to target the MDM2 E3 ligase that is overexpressed in liver cancer [312]. Forth this, attention needs to be directed towards the selection of linker which connects two functional heads: a ligand for E3 ligase recognition and a ligand for target protein recognition. Linker is also a crucial element of the structural template of PROTACS and plays a key role in efficient ubiquitination of the target protein and its ultimate degradation. The available literature reveals that linker represent the most variable vector of this model and researcher have exploited diverse chemical functionalities as linkers (synthetically tractable alkyl chains, polyethylene glycol, heteroaryl linkers generated via click chemistry) to afford the formation of the PROTAC ternary complexes. The chemist usually varies the length of the linker to fine tune the distance between two participating partner proteins in pursuit of attaining efficient degradation, thereby extracting enhanced therapeutic benefits. To add on, maximal binding affinity can also be attained by optimizing the linker position to afford various derivatizations. It is noteworthy to mention that the linker part is optimized not only for the achieving potent bioactivity but also for rendering appropriate physicochemical properties to the resulting scaffolds. Literature precedents reveals that limited attention has been paid so far towards the linker design, however, the evidenced increasing interest of the researchers towards the design of PROTACS is expected to expand the size of the linker library by exploration of sophisticated functional linkers [309,310,311]. Installation of diverse linkers will enable the chemist to ascertain the impact of subtle and unsubtle structural variations in the chemical nature of linker on the anticancer profile of the PROTACS, ultimately generating the structure activity relationship.

Toolbox of ligands for E3 ligases
Once each component of the PROTAC model is finalized, a site of tetheration on the ligand for the target protein and ligand for E3 ligase is identified either through the previously established structure activity relationship or through the docking study of the ligand with in the active site of target protein. Forth this, a synthetic route is proposed that requires an in-depth knowledge of classical organic chemistry concepts. For the tetheration of components, click chemistry (azide-alkyne cycloaddition), organometallic chemistry (Heck coupling, sonogoshira coupling, stille coupling, suzuki coupling, negishi coupling, Buchwald–Hartwig coupling) and amide coupling methodologies (EDC/HOBT, DCC, TBTU, HBTU, PyBOP mediated) have been most exhaustively utilized. In comparison to other therapeutic modalities, PROTAC offers significant advantages such as elimination of target proteins, good oral availability, ease of attaining high potency/selectivity at nonomolar or even picomolar concentrations and high tissue penetration [309,310,311, 313].
More recently, epigenetic inhibitors have garnered significant attention of researchers in pursuit of degrading the target protein and this has led to the initiation of several explorations for the development of epigenetic inhibitor installed PROTACs (Fig. 10). Some of the promising studies have been covered in this section.

Epigenetic inhibitor based PROTACs
Small-molecule HDAC degraders (PROTAC)
Wu et al. reported multifunctional HDAC6 degraders furnished via tetheration of Nexturastat A, a selective HDAC6 inhibitor, with CRBN ligand. Variation of the linker length as well as the linking position culminated in the series of compounds that were evaluated for their HDAC6 degradation effects along with efficacy in multiple myeloma. The results of the study led to the identification of a PROTAC 75 that demonstrated striking selectivity as well as potency and selectivity for the HDAC6 degradation, clearly evidenced with the upregulated levels of HDAC6. The PROTAC also demonstrated promising antiproliferative effects in multiple myeloma cells [314]. Installation of nexturasat in the PROTAC model was attempted in another study performed by Rao et al. Resultantly, a potent PROTAC 76 was identified that exerted significant reduction in the levels of HDAC6 protein level (HeLa cells) at a concentration of 100 nM [315]. Yang et al. recently reported HDAC6 degraders via recruitment of VHL (ligand for E3 ligase) instead of CRBN and identified an extremely potent PROTAC 77 as HDAC6 degrader. Overall the results of the study were highly optimistic and indicated that PROTAC can be utilized as a specific chemical probe for HDAC6 degradation to further investigate HDAC6-related biological pathways (Fig. 10) [316].
Small-molecule BET degraders (PROTAC)
Previously conducted structure-guided design of [1,4] oxazepines by Qin et al. led to the identification of QCA276 that was used as a starting point for the design of small-molecule BET degraders (PROTAC). The degraders were synthesized and subsequently evaluated for BET degradation. Owing to the exhaustive pharmacological explorations, one of the compound, QCA570 (78) emerged as an extremely potent BET degrader 78 that demonstrated BET proteins degradation along with cell growth inhibitory activity towards human acute leukemia cell lines at low picomolar concentrations. In leukemia xenograft models, complete and durable tumor regression was attained by the use of QCA570 (78) at well-tolerated dose-schedules [317]. Crew et al. reported ARV-771 (79) as a pan-BET degrader that caused degradation of c-MYC and induced apoptosis of cells through PARP cleavage. The degrader was evaluated in vivo employing the VCaP tumor model and it was observed that the degrader could exert significant tumor growth inhibition. On the whole, ARV-771 (79) demonstrated high potential for treating CRPC compared to enzalutamide [318]. Bardners et al. furnished a BET degrader via conjugation of JQ1 and pomalidomide. The resulting degrader 80 displayed substantial degradation potential towards BRD4 and could also inhibit the growth of tumor as evidenced in an in vivo murine hind-limb xenograft model with human MV4-11 leukemia cells [319]. Wang et al. designed a BET degrader 81 using a previously reported BET inhibitor (BETi-211) for the treatment of TNBC. The degrader targeted BRD2, BRD3 and BRD4 in a dose-dependent manner and inhibited a TNBC cell growth at nanomolar concentration. A time dependent downregulation of MCL1 protein was also exerted by the degrader. Moreover, in the patient-derived xenograft model of TNBC, the degrader demonstrated high efficacy [320]. A BRD9 degrader was generated via conjugation of the VHL ligand and a BRD9 inhibitor by cullin et al. The degrader 82 was found to be high efficacious as it could induce the degradation of BRD9 and BRD7 at nonomolar concentrations. The CRBN based PROTAC also displayed remarkable cytotoxic effects against acute myeloid eosinophilic leukemia and malignant rhabdoid tumor [321]. A BRD9 degrader 83 was designed and synthesized by Bradner et al. that could induce the degradation of the target protein at nanomolar concentration and also exhibited more pronounced antiproliferative effects in the human AML MOLM-13 cell line than the inhibitor (BRD 9 inhibitor) used for the construction of the PROTAC assemblage (Fig. 10) [322].
Challenges associated with PROTAC approach
In light of the aforementioned, PROTAC appears to be a fascinating stratagem that holds tremendous potential to save the sinking ships of epigenetic inhibitors particularly the ones that are considered to be failures as single agents and are being only considered as suitable candidates for combination therapy. Albeit, endowed with significant merits, PROTAC approach also poses some challenges that need to be addressed to extract a cancer therapeutic out of it in the longer run. At present, it is reported that an estimated 600 E3 ligases have unique activity profiles and distribution patterns throughout the body, however only a limited number of ligands have been identified for them. Thus an exhaustive screening program is required to be initiated for identifying the ligands for E3 ligases as picking the right ligase to tag the target protein is extremely imperative for the success of this program. Other than this, no obvious explanation has been provided so far in the majority of the reported studies behind the rational for selection of the linker and it appears a random selection of linkers is usually made. In this context, a more pragmatic inclusion of the linkers is required to ascertain conclusive benefits of this approach. In addition, to unleash the true potential of PROTACs, comprehensive explorations are required to be conducted at the clinical level to gain deeper mechanistic insights of the PROTACs that have passed the preclinical stage.
Multitargeting agents
The concept of multitargeting approach is not a new concept and numerous hybrid scaffolds composed of more than one pharmacophore to exert concomitant modulation of two or more targets have been successfully furnished in the last decade. Indeed, epigenetic inhibitors field should be duly credited for the regained attention towards the scaffolds that are promiscuous and were once considered to be “not the first choice” of the medicinal chemist. Usually a biochemical relation between the two targets or the optimistic results demonstrated by a cocktail of drugs lays the foundation for the construction of such scaffolds. Interestingly, this strategy confers a broad platform to the chemist for the generation of logically constructed scaffolds with enhanced antitumor effects. It is noteworthy to mention that this approach has particularly capitalized on the flexibility of the three component HDAC inhibitory pharmacophore and thus the pipeline of multitargeting epigenetic inhibitors is flooded with the agents that inhibit HDAC isoforms along with the other biochemically correlated target. Moreover, the approach has also been extended towards non-epigenetic target and numerous hybrid scaffolds decorated with one fragment from epigenetic inhibitors and the other fragment form epigenetic or non-epigenetic inhibitor are reported. Decades of extensive research indicates that the chemist has displayed utmost proficiency for rationally designing dual epigenetic inhibitory agents and few representative studies that excellently exemplifies this concept are presented below:
Dual HDAC-HSP90 inhibitor
Synergistic anticancer efficacy attained with HDAC and HSP90 inhibition coupled with the evidenced ability of HDAC inhibitors to induce acetylation and inhibit the ATP binding and chaperone function of HSP90 protein provides a strong rationale for the fabrication of dual HDAC-HSP90 inhibitor [323,324,325,326,327,328,329]. To add on, revelations ascertaining that the beneficial effects that can be attained via targeting of the HDAC6/HSP90 Axis in NSCLC [330] further strengthens the logic behind the design of hybrid scaffolds (85) composed of pharmacophoric features of HDAC as well as HSP90 inhibitors. With this background, a hybrid scaffold was furnished bearing the key structural units of a second generation HSP90 inhibitor, AT-13387 (84) and FDA approved hydroxamic acid, SAHA (Fig. 11). The results of the in-vitro and in-vivo studies demonstrated that the resulting adduct (85) could remarkably inhibit the HDAC6 isoform (IC50 = 4.3 nM) and HSP90 protein (IC50 = 46.8 nM) and could also exert substantial antiproliferative effects against the NSCLC (A549 and H1975). Expressions of signatory biomarkers associated with HDAC6 and HSP90 inhibition were also modulated by the hybrid compound (85). Other than the striking in-vitro antiproliferative profile, the hybrid scaffold was also endowed with tumour growth inhibitory potential as evidenced in human EGFR-resistance NSCLC H1975 xenograft model in vivo [331].

Design of dual HDAC-HSP90 inhibitor
Dual HDAC-DNMT inhibitors
Motivated by the optimistic results attained with a cocktail of DNMT and HDAC inhibitor in context of the anticancer efficacy including the suppression of the tumorigenicity of cancer stem-like cells and enhancing cancer immune therapy [332,333,334,335,336], Yuan et al. designed and synthesized a dual DNMT and HDAC inhibitor C02S (87) (Fig. 12) that demonstrated significant enzymatic inhibitory activities against DNMT1, DNMT3A, DNMT3B and HDAC1. It was quite evident from the results that the hybrid compound could also inhibit DNMT and HDAC at cellular levels via inversion of mutated methylation and acetylation and increased expression of tumor suppressor proteins. Detailed investigation of C02S revealed that it could induce reexpression of p16, p21 and TIMP3 and cause DNA damages, modulate multiple cancer hallmarks simultaneously and exert tumor growth suppression in mouse breast cancer models [337].

Design of dual HDAC-DNMT inhibitor
Dual HDAC-LSD1 inhibitors
Literature precedents underscores the association of gene expression silencing with HDAC1/2 and LSD1 enzymatic activities within the CoREST complex (HDAC complex that includes HDAC1, HDAC2, the scaffolding protein CoREST, and LSD1) that contributes to cancer and other diseases [338, 339]. These notions spurred a group of researchers to design dual LSD1/HDAC inhibitors (Fig. 13) anticipating that such constructs might demonstrate enhanced activity coupled with an improved therapeutic window. The results of the study led to the identification of a dual inhibitor, corin, endowed with magnificent anti-proliferative activity against several melanoma lines and cutaneous squamous cell carcinoma lines. It is noteworthy to mention that the dual LSD1/HDAC inhibitors displayed more pronounced efficacy than its parent monofunctional inhibitors. Detailed investigation of corin (89) revealed that its striking pharmacological profile relied on an intact CoREST complex. In the melanoma mouse xenograft model, treatment with corin (89) resulted in slowing of the tumor growth [340].

Design of dual HDAC-DNMT inhibitor
Dual HDAC-BET protein inhibitors
In light of role of BET and HDAC proteins as central regulators of chromatin structure and transcription coupled with the evidenced efficacy attained with the combined BET and HDAC inhibition in pancreatic ductal adenocarcinoma at the preclinical level [341], a hybrid scaffold composed of the structural features of a BET inhibitor ( +)-JQ1 and class I HDAC inhibitor CI994 was generated (Fig. 14). Remarkable tumor cell proliferation was achieved with the hybrid compound in comparison to ( +)-JQ1, CI994 alone or combined treatment of both inhibitors. The hybrid scaffold (91) demonstrated more pronounced inhibition of HDAC1 isoform and retained a similar inhibitory potency against BRD4 bromodomains as that of ( +)-JQ1. [342]

Design of dual HDAC-BET inhibitor
Dual HDAC-EZH2 inhibitors
Previous reports reveal that EZH2 works in tandem with HDACs in the same protein complex and mediates gene transcription repression by increasing histone H3 Lys [343,344,345,346,347,348]. Owing to this functional link, design of dual EZH2/HDAC inhibitor has been conceived as a rational approach to control a number of epigenetic-dependent carcinogenic pathways. In an attempt to capitalize on this useful information, a group of researchers led by Romanelli et al. designed a first-in-class dual EZH2/HDAC inhibitor (93) (Fig. 15) that displayed a balanced inhibitory potential towards both the targets and also inhibited the proliferation of U937, THP1 (hematological malignancies) RH4 (rhabdomyosarcoma), SH-N-SK (neuroblastoma) and U87 (glioblastoma) cancer cell lines. Moreover, in U937 and RH4 cells, the dual inhibitor caused cell cycle arrest in the subG1 phase, induced apoptosis and increased the expression of cell differentiation markers [349].

Design of dual HDAC-EZH2 inhibitor
Dual HDAC-PI3K inhibitor
In pursuit of attaining synergistic effects from simultaneous inhibition of PI3K and HDAC, Thakur et al. designed quinazolin-4-one based hydroxamic acids using a rational approach for the tetheration of the pharmacophores of both the inhibitors (Fig. 16). Resultantly, some of the hybrid adducts were found to be potent as well as selective against selective against PI3Kγ, δ and HDAC6 enzymes. The dual inhibitors also exhibited cell growth inhibitory effects exhibited against multiple cancer cell lines. One of the most promising dual inhibitor induced necrosis in several mutant and FLT3-resistant AML cell lines and primary blasts from AML patients and was not found to be toxic. The hybrid compound (95) was also endowed with a good pharmacokinetic profile when evaluated in mice via imp administration. [350]

Design of dual HDAC-PI3K inhibitor
Dual HDAC-tubulin
An investigation conducted for evaluating the efficacy of vincristine (microtubule destabilizing agent) and vorinostat (HDAC inhibitor) reported that the cocktail of the aforementioned drugs demonstrates synergistic antitumor effects in vitro and in vivo. These promising results were basically attributed to the alteration of the microtubules dynamics via vorinostat exerted HDAC inhibition [351]. Motivated by these findings, dual inhibitors of tubulin polymerization and HDAC were designed by Lamaa et al.employing the key pharmacophoric features of 1,1-diarylethylenes (isoCA-4) and belinostat (Fig. 17). Subsequent evaluation of the dual inhibitors revealed that two of the inhibitors (98 and 99) were endowed with striking antiproliferative activity mediated through substantial inhibition of tubulin polymerization as well as HDAC8. One of the compounds, 99, could induce cell cycle arrest of cancer cells at the G2/M phase via disruption of microtubule organization. Furthermore, docking study also rationalized the binding of these hybrid molecules with both tubulin and HDAC active sites. The compound 99 also exhibited cell growth inhibitory effects against tumoral cell lines such as K562, PC3, U87, and BXPC3 and was also active against the CA-4 refractory human colon adenocarcinoma cell line HT-29 and possessed acceptable physicochemical properties [352].

Design of dual HDAC-tubulin inhibitor
The aforementioned studies perspicuously highlight that this approach holds substantial promise and optimism and its implementation is expected to be continued to shoulder the progress of new scaffolds as inhibitors of the epigenetic targets. It is evident that single molecule multiple targets”, “multiple ligands” or “hybrid” anticancer agents can enhance efficacy and lower drug resistance as multiple cross talks between the signaling networks are involved in cancer. The dual inhibitors also score over the strategy of using the cocktail of drugs (combination therapy) as they eliminate the need of extensive investigations such as dose limiting toxicity, drug-drug interactions, pharmacokinetics, bioavailability, establishment of the dosage regimen that, as such, are required to be conducted for combination therapy [353]. Endowed with the benefits of lowering the risk of possible drug interactions, simplified drug metabolism, improved drug transport and reduced drug R&D costs, it is anticipated that the multitargeting agents might outshine the candidature of combination therapy for the treatment of cancer. The main challenge in front of the medicinal chemist is to furnish assemblage that can exert balanced modulation of both the targets to produce synergistic antiproliferative effects and this can only be accomplished via careful selection of targets, pharmacophores as well as the site of tetheration.
Isoform selective inhibitors: Employing a structural template to furnish selective isoform inhibitors of the enzymes
Selecting an epigenetic target for exploration of chemical entities only leads to partial accomplishment of the task as there is still a galactic challenge in front of the medicinal chemist to design isoform selective inhibitors of the enzyme. In general, selective isoform inhibitors are conceived to be less toxic and more efficacious than the non-selective ones. In this context, the ongoing wave in the field of epigenetic drug discovery filed is heavily inclined towards the construction of selective isoform inhibitors to attain anti-tumor effects. This scenario can be best explained by the consideration of the recent trends in the HDAC inhibitors field. As such, HDAC inhibitors garnered limelight with the discovery of pan-HDAC inhibitors namely vorinostat, romidepsin, belinostat, and panobinostat that were later approved by FDA for treatment of diverse malignancies [67,68,69,70]. Despite the clinical success evidenced with the pan HDAC inhibitors, their use has been associated with some side effects, such as fatigue, diarrhea, nausea, QTc-interval prolongation and thrombocytopenia [354,355,356,357]. These disappointing revelations spurred the researchers to draw their attention towards selective inhibitors of HDAC isoforms. Attaining isoform selectivity via structural engineering approaches appears to be extremely feasible for the HDAC inhibitors in light of their flexible and modular structural template composed of three parts: CAP-Linker-Zinc binding motif. The efforts invested in the past in this context have been delved into the below mentioned categories and some selected examples are discussed.
CAP modification The modular nature of the HDAC inhibitory pharmacophore allows the chemist to fine tune each component to attain an inhibitor with a selective isoform inhibitory profile. For instance, a cap construct tolerates a wide variety of modifications such a placement of aryl/heteroaryl ring (bicyclic/tricyclic as well as fused rings), cycloalkanes, bridgehead adducts, spirocyclic rings, steroidal/terpenoidal framework and so on. Other than the placement of diverse scaffolds as CAP component, literature survey indicates that the chemist has also employed several strategies to modify the cap construct such as structural simplification approach, site translocation and CAP rigidification or confernment of some flexibility to the surface recognition part (CAP). Recently, one of the study conducted in our laboratory attempted lead modification of MS-275 employing a CAP rigidification approach that culminated in the identification of a potent compound that was not only a more potent inhibitor of the class I HDACs but was also endowed with substantial antiproliferative effects against TNBC (Fig. 18) [358].

CAP rigidification approach
It is noteworthy to mention that the CAP component remains to be the most comprehensively explored part of the HDAC inhibitory pharmacophore not just in pursuit of attaining isoform selectivity but also for induction or amplification of antitumor effects. The HDAC inhibitory pipeline, at present, is also endowed with some candidates that are potent as well as selective isoform inhibitors of the enzyme but are unable to produce anti proliferative effects. Tubastatin, a highly selective HDAC6 inhibitor, exemplifies one of such case where the selective HDAC6 inhibition is just able to induce neurodegenerative effects irrespective of that fact that HDAC6 isoform is overexpressed in various malignancies. In this context, a study was conducted by our research group to modify the cap construct of tubastatin (structure simplification approach) and the resulting compounds displayed excellent activity profile against multiple myeloma coupled with a striking HDAC6 inhibitory profile (Fig. 19) [359]. Thus, the role of CAP construct also appears to the crucial for activating an enzyme inhibitor to demonstrate cellular potency.

CAP modification
The surface recognition part has also demonstrated scope for structural alteration to afford a transposition in the pattern of inhibitory effects from pan-HDAC inhibition to selective isoform inhibitors of the enzyme. For example, a study focusing on the structural alteration of PXD-101 reported that conferring few degrees of rigidity to the surface recognition part (CAP) via placement of bicyclic ring (Azaindoles) in the chemical architecture of PXD-101 instilled a transposition in inhibitory effects from pan-HDAC inhibition to selective HDAC6 inhibition. (Fig. 20) [360]

CAP modification
CAP and Linker modification On similar lines, the linker part has also demonstrated significant accommodative ability and several functionalities have been employed for the design of HDAC inhibitor. Digging the structural architecture of FDA approved inhibitors viz. Vorinostat, belinostat and panabinostat reveals that each of the structure comprises of different type of a linker (long chain alkyl in SAHA, benzyl acrylamide in LBH-589 and benzenesulfonyl acrylamide in PXD-101) yet exerts pan-HDAC inhibition. However, utilization of the same linker of SAHA with an altered CAP construct (chemically bulky) leads to two selective HDAC6 inhibitors namely ACY-1215 [361] and Tubacin [362]. The identification of ACY-1215 and tubacin presents only one example but the fact that diverse combinations of CAP and linkers can be exploited to extract selective isoform inhibition is supported by a plethora of studies. A relative comparison of the template of SAHA with tubacin and ACY-1215 makes it evident that expanding the size of the CAP construct can induce a transposed pattern of HDAC enzyme inhibition from pan HDAC inhibition to selective HDAC6 inhibition. Likewise, taking cognizance of the structural features of another highly potent and selective HDAC6 inhibitor, Tubastatin [363], it can be conceived that a relatively rigid and fused CAP construct requires a change in the chemical nature of linker to exert selective HDAC 6 inhibition (a relative comparison of tubastatin with tubacin and ACY-1215) (Fig. 21).

Clinically and preclinically active HDAC inhibitors
Several structure alteration programs on a lead compound (already reported selective HDAC6 inhibitors) have also been conducted in the past where modifications of cap and linker part were concomitantly attempted. A recent lead modification study on tubastatin excellently exemplifies this case as the rigid CAP construct of tubastatin was modified into ring opened indole ring bearing a flexible dimethyl amino substituent via a structural simplification approach and acrylamide unit as present in FDA approved agents PXD-101 and LBH-529 was installed in the linker region. The results of the study were overwhelmingly positive as the structurally modified tubastatin analogs exhibited significant cellular growth inhibitory effects and maintained their tendency to exert preferential HDAC6 inhibition (Fig. 22).

Ring opening strategy
C. Modification at the zinc binding motifs Like the CAP construct and the linker part, selection of the zinc binding motif is also an area that needs tantamount attention to attain selective inhibition of HDAC isoforms, thereby extracting the anticancer effects in malignancies having overexpressed pattern of those isoforms. Practically, the competition is majorly between two classes of zinc binding motif viz. hydroxamic acid and aminoanilides. As such, the former class clearly outshines the latter in terms of clinical success with 3 hydroxamic acid type HDAC inhibitors receiving FDA approval for use in cancer [68,69,70]. However, chidamide represents the only aminoanilide that has been approved by CFDA to treat patients with recurrent or refractory PTCL [72]. Regardless of the clinical success, the present trend in this field is equally aligned towards the development of both the classes of inhibitors. An obvious explanation to this is the evidenced susceptibility of hydroxamic acids to glucuronide conjugation leading to inactivation and off-targeting whereas some explorations reported that aminoanilides are less prone to glucuronide metabolism and are endowed with high efficacy possibly due to their slow, tight binding and slow disassembling with HDACs [190, 191, 363,364,365,366,367,368]. Another notable variation between the two classes can be observed in their enzymatic inhibitory profile as pan HDAC inhibition and selective HDAC 6 inhibition is mostly exerted via use of hydroxamic acid based HDAC inhibitors while class I HDAC selectivity is usually attained via the fabrication of aminoanilides. Other than these two types of zinc binding motifs, a trifluoromethyloxadiazolyl moiety (TFMO) as a non-metal chelating group has further led to a categorical division of HDAC inhibitors as the metal chelators and the non-metal chelators. TFMO interacts by one of the fluorine atoms and its oxygen with the active Zn2 + atom in the catalytic center and represents another class of zinc binding motif [369, 370] (Fig. 23). In light of the debated candidature of hydroxamic acids and to some extent, amino anilides also, due to the susceptibility to glucuronidation based inactivation, explorations have been accelerated for non-metal chelating class of HDAC inhibitors in recent times. As a result, TMP-269 has emerged as a prototype inhibitor that bears a trifluoromethyloxadiazolyl moiety (TFMO) as a nonchelating MBG and acts as a class IIa HDAC selective inhibitor [369]. The attributes of TFMO bearing adducts act as boon for the medicinal chemist working in this area as utilization of this zinc binding motif can accomplish HDAC inhibitors that are non-susceptible to glucoronidation and can generate selective/preferential class IIa inhibitory adducts (class IIa HDAC bias). To add on, leveraging the TFMO group for furnishing the HDAC inhibitory assemblage presents a rational approach that might challenge the strategy of targeting a metalloenzyme only with a metal chelators. It is anticipated that conclusive therapeutic benefits can be achieved by shifting the inclination towards weak binders of the metal that are duly supported by other structural features to garner favourable interactions with the amino acid residues of the active site. This aforementioned information clearly provides a platform to pragmatically design new chemical entities that can target the malignancies having an overexpressed pattern of class IIa HDACs.

Classification on the basis of zinc binding motif
Overall, the approach to construct HDAC inhibitors will require an in-depth literature survey to extract the information regarding the over-expression of various HDAC isoforms in diverse malignancies and accordingly adducts can be designed to bear carefully recruited components of the HDAC inhibitory model. Consideration of these notions can certainly enable us to create a depository of isoform selective HDAC inhibitors that upon further evaluation such as evaluation of ADME properties, high dose pharmacology, toxicity studies, evaluation in animal models that mimics the human condition can advance to clinical stage investigation.
Fragment stitching approach: Fragment stitching on an existing drug to produce more potent derivatives
Unlike the approach of using a structural template to create a compendium of compounds (via diverse permutations and combinations of key components) to target various isoforms of the enzyme, this strategy simply employs a FDA approved drug to stich diverse fragments that either leads to the induction or potentiation of anticancer effects. Usually a transposition in the isoform inhibitory effects is not desired rather inhibition of the same isoform is capitalized to derive antitumour effects in diverse malignancies. This approach can be best explained by the numerous investigations conducted on a LSD1 inhibitor, tranylcypromine, which has been exhaustively utilized in the past to fill the library of rationally designed analogues with anticancer efficacy (Fig. 24). To exemplify this, some selected structures have been presented in Fig. 24 that were generated via stitching of diverse fragments on the tranylcypromine core. Compound 108 was recently reported by Vianello et al. as potent LSD 1inhibitor that displayed substantial activity in the in vivo model (in the murine promyelocytic leukemia model) on oral administration. The results of the study also demonstrated that compound 108 was well tolerated and led to remarkable prolongment of the survival time of the treated mice (35% and 62% at the doses of 11.25 and 22.50 mg/kg, respectively). [371] Rotili et al. conducted an investigation that led to the identification of LSD1 inhibitors (109 and 110). The inhibitors induced an increase in H3K4 and H3K9 methylation levels in cells, caused growth arrest and apoptosis in LNCaP prostate and HCT116 colon cancer cells. [372] A study by gehling et al. led to the identification of a highly potent and selective LSD 1 inhibitor, compound 111, (< 4 nM biochemical, 2 nM cell, and 1 nM GI50). Compound 111 exhibited cell growth inhibitory effects in a panel of AML cell lines along with notable antitumor potential in a Kasumi-1 xenograft model of AML at the dose of 1.5 mg/kg once daily (administered orally) [373].

Fragment stitching approach
The structures shown in Fig. 24 only represents some of the selected examples that have been furnished using the aforementioned approach, however, the preclinical pipeline of LSD1 inhibitors is endowed with numerous candidates synthesized via fragment stitching approach. It is noteworthy to mention that this approach has been implemented to all classes of epigenetic inhibitors and the main advantage of employing this approach is that substantial amount of information in terms of protocol for synthesis, structure activity relationship as well as the toxicity profile is available and accessible. This information plays a key role in expediting the library generation of compounds and it is conceived that this startegem is likely to be continually employed in the near future to create antitumor assemblages.
Antibody drug conjugates: Accomplished via conjugation of a small molecule inhibitor and a humanized antibody though a chemical linker, ADCs selectively bind to the receptors of tumor cells. Internalization of the receptor–ADC complex usually occurs through the endocytosis pathway. Forth the internalization, the cytotoxic drug is released via cleavage of the linker. This strategy of targeted drug delivery presents enough promise to overcome the issue of systemic toxicity and narrow therapeutic window that limits the clinical use of the available chemotherapeutic agents [374,375,376]. In this context, a study was conducted recently and two antibody-drug conjugates (112 and 113) comprising of a HDAC inhibitor ST7612AA1 and cetuximab employing cleavable and non-cleavable linkers were synthesized. The results of the study were extremely promising and the HDAC inhibitor-antibody conjugate demonstrated efficient internalization in tumour cells. In the in vivo studies, the conjugates exhibited striking antitumor activity (animal models of human solid tumors) without any toxicity that is generally observed with traditional ADCs delivering highly potent cytotoxic drugs. (Fig. 25) [377]

Antibody drug conjugates
Another research group recently furnished an ADC endowed with the potential to deliver a HDAC inhibitor to ErbB2 + solid tumors. In the study, partial reduction of trastuzumab with tris [2-carboxyethyl] phosphine was performed followed by conjugation to ST7464AA1, the active form of the prodrug HDAC inhibitor ST7612AA1 via a maleimide-thiol linker to furnish the target ADC ST8176AA1. The detailed biological evaluation of ST8176AA1 revealed a similar receptor binding and internalization of ST8176AA1 to trastuzumab. However, the conjugate demonstrated higher anti-tumor activity compared to trastuzumab (in vitro assay) that correlated with increased acetylation of histones and α-tubulin. Moreover, similar to trastuzumab, ST8176AA1 also increased the expression of ErbB2 and estrogen receptor in TNBC cells. (Fig. 25) [378]
These aforementioned studies provide a strong rational to extend this concept to the targeted delivery of epigenetic inhibitors that, in turn, can demonstrate fruitful results in cancer by exerting epigenetic modulation at a much safer dose in comparison to standard epigenetic inhibitors based therapeutic tools. In this context, it appears logical to dig the preclinical pipeline of the epigenetic inhibitors and select some candidates that could not advance to the clinical stage investigation owing to the toxicity related hindering factors. Once selected, their accommodation in ADC model should be attempted.
(CRISPR) Cas9
Designing the CRISPR/Cas9-based strategies to target the cancerous epigenetic regulators in a more specific manner is an emerging potential approach that has garnered tremendous interest in the recent past as a tool to correct genetic mutations. CRISPR-based epigenome editors (CRISPR epi-editors) consist of dCas9 and epigenetic effector (fused or non-covalently) [379,380,381,382] and are being given serious consideration as a practical approach in cancer gene therapy as they can activate the tumor suppressor genes and also inhibit the tumor driving genes. On the literature precedential basis, it is well known that epigenetic mechanisms sometimes inactivate tumor suppressors in some cancers. To add on, the epigenetic factors, such as LSD1, EZH2 and NSD2 (tumor drivers,) are overexpressed by either epigenetic or genetic mechanisms in diverse malignancies. In light of these revelations, development of CRISPR/Cas9 based transcriptional regulators appears to be a pragmatic way to: (1) suppress the expression of the aforementioned enzymes in some cancers (2) target the driver genes of cancer as well as the genes essential for cancer maintenance or drug resistance (3) target the “undruggable” epigenetically silenced tumor suppressors [379,380,381,382,383]. In a nutshell, dCas9-fused epigenetic regulators holds enough promise for cancer treatment as they can reversibly manipulate the epigenetic patterns and also regulate the oncogenes and tumor suppressors expressions.
Conclusion and future perspective
Significant advancement has been made in the field of the epigenetic drug discovery in the last decade. All the major classes of the epigenetic tools have received considerable attention. In context of the DNMT inhibitors, the FDA approved DNMT inhibitors 5-azacytidine and decitabine were exhaustively explored in AML and MDS at the clinical level and optimistic results were attained particularly in combination therapy. Other than the FDA approved nucleoside based DNMT inhibitors, guadecitabine, a dinucleotide antimetabolite of a decitabine linked via phosphodiester bond to guanosine and 5-fluoro-2′-deoxycytidine (FdCyd) also demonstrated promise and are likely to be the subjects of number of investigations in diverse cancer types in the near future. In addition, non-nucleoside prototypes namely curcumin, hydralazine, procainamide, disulfiram, SGI-1027 and ((-)-epigallocatechin-3-gal were evidenced to be endowed with interesting antitumor profiles. In light of the current trends in medicinal chemistry, it appears that the balance in context of the future research will be tilted towards the natural product based non-nucleoside inhibitors owing to the beneficial trends evidenced with curcumin coupled with the fact that the chemical architecture of this phytoconstituent comprises of several sites accessible to generate an appropriate sized structure pool. For the EZH2 inhibitory field, several drug candidates are undergoing clinical stage evaluation with GSK126, GSK343, CPI-205, ZLD1039, PF-06821497, UNC1999 representing a few of them. It is noteworthy to mention that the clinical success of tazemetosat has brought the spotlight on EZH2 as a potential target for the design of cancer therapeutics. The studies conducted also indicates that significant benefits can be attained through the simultaneous inhibition of EZH1/EZH2 and thus the dual targeting of the aforementioned is likely to be explored more comprehensively. Moreover, the synergistic effects anticipated to be attained through the combination of EZH2 inhibitors with immunotherapy or chemotherapy are also likely to be evaluated. In context of the preliminary/preclinical exploration, the chemical architectures of tazemetostat offer several sites for chemical alterations that can be exploited to create library of new inhibitors. The area of development of DOT1L inhibitors have relatively received less attention in comparison to the other epigenetic tools and pinometostat, a potent and selective small molecule DOT1L inhibitor, stands as the only DOT1L inhibitor that has been a subject of significant clinical attention. In this context, there is an urgent need to load the armoury of the DOT1L inhibitors with new entries that can maximize the therapeutic benefits of inhibiting this epigenetic target in cancer. HDAC inhibitors as small molecule inhibitors have always been at the forefront of investigations and a number of inhibitors belonging to this class of hydroxamic acids and anilides were tested in diverse malignancies both as single agents as well as the part of combination regimens. While these two classes are expected to continue being evaluated clinically as well as preclinically, it is also anticipated that some initiatives will be taken to probe the candidature of the non-metal chelating class of HDAC inhibitors as a potential therapeutic weapon in cancer. A complete profiling of TMP-269, prototype non-metal chelating HDAC inhibitor, might open an avenue for the construction of similar scaffolds. As the non-metal chelating class is expected to be relatively free of the pharmacological liabilities of the metal chelating class of HDAC inhibitors, safer therapeutics can be established via furnishment of agents that binds weakly with the metals but relies on interactions from other parts of the pharmacophore to afford acceptable levels of isoform inhibition. Progress in the field of LSD1 inhibitors has basically relied on the fragment stitching approach on the chemical architecture of tranylcypromine to access new frameworks capable of producing antiproliferative effects. OPY-1001, IMG-7289, GSK-2879552 and ORY-2001 are the tranylcypromine based LSD1 inhibitors that are undergoing clinical stage investigation in different cancer types. Futuristic attempts are likely to be directed towards the hydrazide type LSD1 inhibitors owing to the striking antitumor potential demonstrated by SP-2509. OTX-015, CPI-0610, BMS-986158, GSK525762, INCB054329, INCB057643, ODM-207, RO-6870810, BAY1238097, CC-90010, AZD-5153, PLX-51107, SF-1126 represents the BET inhibitors in various phases of clinical evaluation. It is quite hopeful that a comprehensive profiling of these BET inhibitors might yield a cancer therapeutic in the longer run. In addition, some preclinical studies have excellently utilized the BET inhibitory scaffolds for installation in the PROTACS model. The optimistic activity profile of the BET inhibitor based degraders can also spur numerous investigations in this direction. To sum up, the armoury of clinical and preclinical epigenetic inhibitors is filled with numerous candidates, however, conclusive benefits can only be ascertained after the completion of all the phases of the clinical trials.
Overall, the findings covered in this perspective present recent advances in the field of epigenetic drug discovery. It is apparent that future research will include parallel programs aimed at the completion of the ongoing clinical studies, initiation of new clinical trials and development of new inhibitors. In addition, numerous brain storming sessions involving expertise of interdisciplinary teams composed of chemist, biologist, and formulation chemist along with researchers well versed with computational aspects of drug design needs to be conducted. Such programs can identify the hurdles and the hindering factors that have halted the clinical growth of several inhibitors of the epigenetic targets and present logical ways to strengthen the candidature of epigenetic targets in context of capacity to generate single target agents.
Availability of data and materials
Not applicable.
Abbreviations
- PROTACS:
-
Proteolysis targeting chimera
- DNMT:
-
DNA methyltransferase
- HDAC:
-
Histone deacetylase
- LSD:
-
Lysine specific demethylase
- EZH2:
-
Enhancer of zeste homolog 2
- BET:
-
Bromodomain and extra-terminal motif
- DOT1L:
-
Disruptor of telomeric silencing 1-like
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- HMT:
-
Histone methyl transferase
- miRNAs:
-
MicroRNAs
- TSG:
-
Tumor suppressor genes
- HSP:
-
Heat shock protein
- ADC:
-
Antibody drug conjugate
- FDA:
-
Food and drug administration
- CR:
-
Complete response
- PR:
-
Partial response
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- CI:
-
Confidence interval
- MF:
-
Myelofibrosis
- HCC:
-
Hepatocellular carcinoma
- CRi:
-
Incomplete blood count recovery
- CRp:
-
Incomplete platelets recovery
- PRC2:
-
Polycomb repressive complex
- RP2D:
-
Recommended phase 2 dose
- AE:
-
Adverse events
- MTD:
-
Maximum tolerated dose
- MLL:
-
Mixed lineage leukemia
- R/R:
-
Relapsed/refractory
- SD:
-
Stable disease
- CTCL:
-
Cutaneous T-Cell lymphoma
- DLBCL:
-
Diffuse B-cell lymphoma
- HAT:
-
Histone acetyltransferase
- DLT:
-
Dose limiting toxicity
- NSCLC:
-
Non SCLC
- TCL, PTCL:
-
PTCL
- CML:
-
Chronic myelogenous leukemia
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- OR:
-
Objective response
- CRPC:
-
Castration-resistant prostate cancer
- SCLC:
-
SCLC
- NHL:
-
Non-Hodgkin’s lymphoma
- HER2−:
-
Human epidermal growth factor receptor 2 negative
- HR+ :
-
Hormone receptor-positive
- TAAEs:
-
Treatment associated adverse events
- ADC:
-
Antibody drug conjugates
- KDMs:
-
Histone demethylase
- SAM:
-
S-adenosyl-methionine
- DCR:
-
Disease control rate
- CD:
-
Cytidine deaminase
- CTC:
-
Circulating tumor cell
- MLL-r:
-
MLL rearrangements
- HL:
-
Hodgkin’s lymphoma
- CBR:
-
Clinical benefit rate
- NMPA:
-
National medical product administration
- MLFS:
-
Morphological leukemia-free state
- NMC:
-
NUT midline carcinoma
- TNBC:
-
Triple negative breast cancer
References
Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Gen. 2018;19:371–84.
Bennet RL, Licht JD. Targeting epigenetics in cancer. Ann Rev Pharm Toxicol. 2018;58:187–207.
Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. 2018;378:323–1334.
Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods Mol Biol. 2015;1238:3–25.
Kondo Y, Katsushima K, Ohka F, Natsume A, Shinjo K. Epigenetic dysregulation in glioma. Cancer Sci. 2014;105:363–9.
Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8.
Maze I, Noh KM, Allis CD. Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacol. 2013;38:3–22.
Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis.” PNAS. 2006;103:6428–35.
O’Donnell KJ, Meaney MJ. Epigenetics, development, and psychopathology. Ann Rev Clin Psychol. 2020;16:327–50.
Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell. 2013;154:490–503.
Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50.
Stilling RM, Fischer A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:19–26.
Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107:22687–92.
Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–23.
Liu R, Lei JX, Luo C, Lan X, Chi L, Deng P, et al. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2012;45:902–12.
Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18:1225–34.
Zhang Z, Schluesener YHJ. Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J Neuropathol Exp Neurol. 2013;72:178–85.
Miller AA, Kurschel E, Osieka R, Schmidt CG. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur J Cancer Clin Oncol. 1987;23:1283–7.
Chen KL, Wang SS, Yang YY, Yuang RY, Chen RM, Hu CJ. The epigenetic effects of amyloidbeta (1–40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Comm. 2009;378:57–61.
Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, Kirsch W. Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics. 2012;12:1261–8.
Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, et al. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J Alzheimer Dis. 2009;18:131–9.
Ganesan A, Arimondo PB, Rots MG, Jeronimo C, Berdasco M. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenet. 2019;11:174.
Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer. 2011;11:726–34.
Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20:109–27.
Lamb YN, Dhillon S. Epi proColon® 20 CE: A blood-based screening test for colorectal cancer. Mol Diagn Ther. 2017;21:225–32.
Ned RM, Melillo S, Marrone M. Fecal DNA testing for colorectal cancer screening: the ColoSureTM test. PLoS Curr. 2011;3:RRN1220.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92.
Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455.
Bishop KS, Ferguson LR. The interaction between epigenetics, nutrition and the development of cancer. Nutrients. 2015;7:922–47.
Supic G, Jagodic M, Magic Z. Epigenetics: a new link between nutrition and cancer. Nut Cancer. 2013;65:781–92.
Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol. 2009;625:131–42.
Holliday R. DNA methylation and epigenetic inheritance. Philos Trans R Soc Lond B Biol Sci. 1990;326:329–38.
Pechalrieu D, Etievant C, Arimondo PB. DNA methyltransferase inhibitors in cancer: from pharmacology to translational studies. Biochem Pharmacol. 2017;129:1–13.
Verma M, Rogers S, Divi RL, Schully SD, Nelson S, Su JL, et al. Epigenetic research in cancer epidemiology: trends, opportunities, and challenges. Cancer Epidemiol Biomark Prevent. 2014;23:223–33.
Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698–711.
Wild L, Flanagan JM. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim Biophys Acta. 2010;1806:50–7.
Wang LH, Wu CF, Rajasekaran N, Shin YK. Loss of tumor suppressor gene function in human cancer: an overview. Cell Phys Biochem. 2018;51:2647–93.
Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–52.
Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202.
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49.
Marmorstein R. Structure of histone acetyltransferases. J Mol Biol. 2001;311:433–44.
Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–58.
Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–89.
Jia SWYu. Degrees make all the difference. Epigenetics. 2009;4:1–4.
Gilbert SF. Developmental biology. 6th edition. Sunderland (MA): Sinauer Associates; 2000. https://www.ncbi.nlm.nih.gov/books/NBK9983/.
Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–98.
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53.
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–6.
Albert M, Helin K. HMT in cancer. Semin Cell Dev Biol. 2010;21:209–20.
Cieśla J, Frączyk T, Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol. 2011;58:137–48.
Johnson LN, Barford D. The effects of phosphorylation on the structure and function of proteins. Annu Rev Biophys Biomol Struct. 1993;22:199–232.
Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–30.
Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Dis. 2002;4(4):309–15.
Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2011;411:355–65.
Ubersax JA, Ferrell JE Jr. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Bio. 2007;8:30–541.
Fabbro D, Cowan-Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR review 14. Br J Pharmacol. 2015;172:2675–700.
Kimura Y, Tanaka K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. J Biochem. 2010;147:793–8.
Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428.
Gill G. Something about SUMO inhibits transcription. Curr Opin Genet Dev. 2005;15:536–41.
Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17:184.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–9.
Malumbres M. miRNAs and cancer: an epigenetics view. Mol Aspects Med. 2013;34:863–74.
Galasso M, Sana ME, Volinia S. Non-coding RNAs: a key to future personalized molecular therapy? Genome Med. 2010;2:12.
Benetatos L, Vartholomatos G, Hatzimichael E. MEG3 imprintedgene contribution in tumorigenesis. Int J Cancer. 2011;129:773–9.
Mei YP, Liao JP, Shen J, Yu L, Liu BL, Liu L, et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene. 2012;31:2794–804.
Liu NA, Zhao R, Ma Y, Wang D, Chen Y, Zhou D, et al. The development of epigenetics and related inhibitors for targeted drug design in cancer therapy. Curr Top Med Chem. 2018;18:2380–94.
Barbarotta L, Hurley K. Romidepsin for the treatment of peripheral T-celllymphoma. J Adv Pract Oncol. 2015;6:22–36.
Marks A. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6.
Qian G, Ara G, Mills E, LaRochelle WJ, Lichenstein HS, Jeffers M. Activity of the histone deacetylase inhibitor belinostat (PXD101) in preclinical models of prostate cancer. Int J Cancer. 2008;122:1400–10.
Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res. 2015;21:4767–73.
FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-option-specifically-patients-epithelioid-sarcoma-rare-soft-tissue. Accessed 18 Jan 2020.
Guan XW, Wang HQ, Ban WW, Chang Z, Chen HZ, Jia L, et al. Novel HDAC inhibitor Chidamide synergizes with Rituximab to inhibit diffuse large B-cell lymphoma tumour growth by upregulating CD20. Cell Death Dis. 2020;11:1–18.
Gius D, Cui H, Bradbury CM, Cook J, Smart DK, Zhao S, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 2004;6:361–71.
Liu N, Zhao R, Ma Y, Wang D, Yan C, Zhou D, et al. The development of epigenetics and related inhibitors for targeted drug design in cancer therapy. Curr Top Med Chem. 2018;18:2380–94.
Kelly KT, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–78.
Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Therapy. 2019;4:62.
Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95:399–409.
Presant CA, Coulter D, Valeriote F, Vietti TJ. Contrasting cytotoxicity kinetics of 5-azacytidine and dihydro-5-azacytidine hydrochloride in L1210 leukemia in mice. J Natl Cancer Inst. 1981;66:1151–4.
Fenaux P, Mufti GJ, Lindberg EH, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk MDSs: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32.
Chou WC, Yeh SP, Hsiao LT, Lin SF, Chen YC, Chen TY, et al. Efficacy, safety, and pharmacokinetics of subcutaneous azacitidine in Taiwanese patients with higher-risk MDSs. Asia Pac J Clin Oncol. 2017;13:e430–9.
Seymour JF, Döhner H, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. Azacitidine improves clinical outcomes in older patients with acute myeloid leukaemia with myelodysplasia-related changes compared with conventional care regimens. BMC Cancer. 2017;17:852.
Krug U, Koschmieder A, Schwammbach D, Gerss J, Tidow N, Steffen B, et al. Feasibility of azacitidine added to standard chemotherapy in older patients with AML—a randomised SAL pilot study. PLoS ONE. 2012;7:e52695.
Ohanian M, Garcia-Manero G, Levis M, Jabbour E, Daver M, Borthakur G, et al. Sorafenib combined with 5-azacytidine in older patients with untreated FLT3-ITD mutated AML. Am J Hematol. 2018;93:1136–41.
Stevens B, Winters A, Gutman JA, Fullerton A, Hemenway G, Schatz D, et al. Sequential azacitidine and lenalidomide for patients with R/R AML: clinical results and predictive modeling using computational analysis. Leuk Res. 2019;81:43–9.
Medeiros BC, McCaul K, Kambhampati S, Pollyea DA, Kumar R, Silverman LR, et al. Haematologica. 2018;103:101–6.
Aedes L, Recher C, Lejeune J, Stamatoullas A, Sebert M, Delaunay J, et al. A phase I–II study of the efficacy and safety of lenalidomide (LEN) combined to azacitidine (AZA) in higher risk MDS and AML with Del 5q—a study by the groupe francophone des myelodysplasies (GFM). Blood. 2015;126:2892–2892.
Guillaume T, Malard F, Magro L, Labopin M, Tabrizi R, Borel C, et al. Prospective phase II study of prophylactic low-dose azacitidine and donor lymphocyte infusions following allogeneic hematopoietic stem cell transplantation for high-risk AML and MDS. Bone Marrow Transplant. 2019;54:1815–26.
Yalniz FF, Daver N, Rezvani K, Steven KS, Maro O, Borthakur G, et al. A pilot trial of lirilumab with or without azacitidine for patients with MDS. Clin Lymphoma Myeloma Leuk. 2018;18:658-663.e2.
Strati P, Kantarjian H, Ravandi F, Nazha A, Borthakur G, Daver N, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with AML and MDS. Am J Hematol. 2015;90:276–81.
Holsinger HL, Ramakrishnan A, Storer M, Becker PS, Petersdorf S, Deeg HJ, et al. Therapy of MDS (MDS) with azacitidine given in combination with etanercept: a phase II study. Blood. 2007;110:1452.
Von Hoff DD, Rasco DW, Heath EI, Munster PN, Schellens JHM, Isambert N. Phase I study of CC-486 alone and in combination with carboplatin or nab-paclitaxel in patients with R/R solid tumors. Clin Cancer Res. 2018;24:4072–80.
Dickinson M, Cherif H, Fenaux P, Mittelman M, Verma A, O Portella MS, et al. Azacitidine with or without eltrombopag for first-line treatment of intermediate- or high-risk MDS with thrombocytopenia. Blood. 2018;132:2629–38.
Masarova L, Verstovsek S, Hidalgo-Lopez JE, Pemmaraju N, Bose P, Estrov Z, et al. A phase 2 study of ruxolitinib in combination with azacitidine in patients with MF. Blood. 2018;132:1664–74.
U.S. Food and Drug Administration Approves Onureg® (azacitidine tablets), a new oral therapy, as continued treatment for adults in first remission with AML. https://www.businesswire.com/news/home/20200901005914/en/U.S.-Food-and-Drug-Administration-Approves-Onureg%C2%AE-azacitidine-tablets-a-New-Oral-Therapy-as-Continued-Treatment-for-Adults-in-First-Remission-with-Acute-Myeloid-Leukemia. Accessed 31 Nov 2020.
Beisler JA, Abbasi MM, Kelley JA, Driscoll JS. Synthesis and antitumor activity of dihydro-5-azacytidine, a hydrolytically stable analogue of 5-azacytidine. J Med Chem. 1977;20:806–12.
Presant CA, Coulter D, Valeriote F, Vietti TJ. Contrasting cytotoxicity kinetics of 5-azacytidine and dihydro-5-azacytidine hydrochloride in L1210 leukemia in mice. J Natl Cancer Inst. 1981;66:1151–4.
Issa JPJ, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23:3948–56.
Blum W, Sanford BL, Klisovic R, DeAngelo DJ, Uy G, Powell BL, et al. Maintenance therapy with decitabine in younger adults with AML in first remission: a phase 2 cancer and leukemia Group B Study (CALGB 10503). Leukemia. 2017;31:34–9.
Oki Y, Jelinek J, Shen L, Kantarjian HM, Issa JPJ. Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood. 2008;111:2382–4.
Jabbour E, Kantarjian H, O’Brien S, Kadia T, Malik A, Welch MA, et al. Retrospective analysis of prognostic factors associated with response and overall survival by baseline marrow blast percentage in patients with MDSs treated with decitabine. Clin Lymphoma Myeloma Leuk. 2013;13:592–6.
Bohl SR, Claus R, Dolnik A, Schlenk RF, Döhner K, Hackanson B, et al. Decitabine response associated gene expression patterns in AML (AML). Blood. 2013;122:3756.
Mawad R, Becker PS, Hendrie P, Scott B, Wood BL, et al. Phase II study of tosedostat with cytarabine or decitabine in newly diagnosed older patients with acute myeloid leukaemia or high-risk MDS. Br J Haematol. 2016;172:238–45.
FDA Approves New Therapy for MDSs (MDS) That Can Be Taken at Home. https://www.fda.gov/news-events/press-announcements/fda-approves-new-therapy-myelodysplastic-syndromes-mds-can-be-taken-home. Accessed 27 Nov 2020.
Guadecitabine (SGI-110). https://astx.com/research-development/clinical-pipeline/guadecitabine-sgi-110-dnmt-inhibitor-solid-tumors/. Accessed 21 Oct 2020.
Chung W, Kelly AD, Kropf P, Fung H, Jelinek J, Su XY, et al. Genomic and epigenomic predictors of response to guadecitabine in R/R acute myelogenous leukemia. Clin Epigenet. 2019;11:106.
A randomized, controlled, open-label, Phase 2 Trial of SGI-110 and carboplatin in subjects with platinum-resistant recurrent ovarian cancer. https://www.clearityfoundation.org/wpcontent/uploads/2016/08/NCT01696032.pdf. Accessed 25 Oct 2020.
Pharmacodynamic (PD) and pharmacokinetic (PK) results of the second-generation hypomethylating agent, SGI-110, in patients with HCC(HCC) after progression on sorafenib. https://astx.com/wp-content/uploads/2016/11//2015_SGI-110_Posterl_AACR_El-Khoueiry_abst-2947_final.pdf. Accessed 23 Oct 2020.
Astex Pharmaceuticals and Otsuka announce results of the phase 3 ASTRAL-1 study of guadecitabine (SGI-110) in treatment-naïve AML patients ineligible to receive intense induction chemotherapy. https://astx.com/astex-pharmaceuticals-and-otsuka-announce-results-of-the-phase-3-astral-1-study-of-guadecitabine-sgi-110-in-treatment-naive-aml-patients-ineligible-to-receive-intense-induction-chemotherapy/. Accessed 26 Sep 2020.
Astex and Otsuka announce results of phase 3 ASTRAL-2 and ASTRAL-3 studies of guadecitabine (SGI-110) in patients with previously treated AML (AML) and MDSs or chronic myelomonocytic leukemia (MDS/CMML). https://astx.com/astex-and-otsuka-announce-results-of-phase-3-astral-2-and-astral-3-studies-of-guadecitabine-sgi-110-in-patients-with-previously-treated-acute-myeloid-leukemia-aml-and-myelodysplastic-syndromes-or/. Accessed 27 Sep 2020.
Holleran JL, Beumer JH, McCormick DL, Johnson WD, Newman EM, Doroshow JH, et al. Oral and intravenous pharmacokinetics of 5-fluoro-2’-deoxycytidine and THU in cynomolgus monkeys and humans. Cancer Chemother Pharmacol. 2015;76:803–11.
Phase I trial of oral 5-fluoro-2′-deoxycytidine with oral tetrahydrouridine in patients with advanced solid tumors. https://reader.elsevier.com/reader/sd/pii/S0959804916328647?token=596E0616E2B0932EE223226E4AB7F7F6E27EC084239D4B2B56DA67BFC58D84C5EC8C9680CD5FAFF23D708C03F4F04308. Accessed 21 Nov 2020.
Newman EM, Morgan RJ, Kummar S, Beumer JH, Blanchard MS, Ruel C, et al. A phase I, pharmacokinetic, and pharmacodynamic evaluation of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine, administered with tetrahydrouridine. Cancer Chemother Pharmacol. 2015;75:537–46.
Sullivan Coyne G, Wang L, Zlott J, Juwara L, Covey JM, Beumer JH, Cristea M, et al. Intravenous 5-fluoro-2′-deoxycytidine administered with tetrahydrouridine increases the proportion of p16-expressing circulating tumor cells in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2020;85:979–93.
Holleran JL, Parise RA, Erin J, Eiseman JL, Covey JM, Glaze ER, et al. Plasma pharmacokinetics, oral bioavailability, and interspecies scaling of the DNA methyltransferase inhibitor, zebularine. Clin Cancer Res. 2005;11:3862–8.
Brueckner B, Boy RG, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65:6305–11.
Piña IC, Gautschi JT, Wang GYS, Sanders ML, Schmitz FJ, France D. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J Org Chem. 2003;68:3866–73.
Fang M, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–70.
Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988;140:2197–200.
Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res. 2003;9:1596–603.
Villar-Garea A, Fraga MF, Espada J, Esteller M. Procaine is a DNAdemethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003;63:4984–9.
Candelaria M, Gallardo-Rincón D, Arce C, Cetina L, Aguilar-Ponce JL, Arrieta O, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007;18:1529–38.
Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med Oncol. 2011;28:540–6.
Davis AJ, Gelmon KA, Siu LL, Moore MJ, Britten CD, Mistry N, et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Investig New Drugs. 2003;21:85–97.
Amato RJ. Inhibition of DNA methylation by antisense oligonucleotide MG98 as cancer therapy. Clin Genitourin Cancer. 2007;5:422–6.
Plummer R, Vidal L, Griffin M, Lesley M, de Bono J, Coulthard S, et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3177–83.
Stresemann C, Brueckner B, Musch T, Stopper H, Lyko F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006;66:2794–800.
Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate. 2011;71:333–43.
Datta J, Ghoshal K, Denny WA, Gamage SA, Brooke DG, Phiasivongsa P, et al. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009;69:4277–85.
Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9.
Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11.
Raman JD, Mongan NP, Tickoo SK, Boorjian SA, Scherr DS, Gudas LJ. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clin Cancer Res. 2005;11:8570–6.
Matsukawa Y, Semba S, Kato H, Ito A, Yanagihara K, Yokozaki H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006;97:484–91.
Sudo T, Utsunomiya T, Mimori K, Nagahara H, Ogawa K, Inoue H, et al. Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br J Cancer. 2005;92:1754–8.
Watanabe H, Soejima K, Yasuda H, Kawada I, Nakachi I, Yoda S, et al. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008;8:15.
Chen Y, Xie D, Li YW, Cheung CM, Yao H, Chan CY, et al. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Canc Lett. 2010;297:109–16.
Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13:104.
Nutt SL, Keenan C, Chopin M, Allan RS. EZH2 function in immune cell development. Biol Chem. 2020;401:933–43.
Yao Y, Hu H, Yang Y, Zhou G, Shang Z, Yang X, et al. Downregulation of enhancer of zeste homolog 2 (EZH2) is essential for the induction of autophagy and apoptosis in colorectal cancer cells. Genes (Basel). 2016;3:10.
Ito T, Teo YV, Evans SA, Neretti N, Sedivy JM. Regulation of cellular senescence by polycomb chromatin modifiers through distinct DNA damage- and histone methylation-dependent pathways. Cell Rep. 2018;22:3480–92.
Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491–5024.
Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–88.
Yap TA, Winter JN, Giulino-Roth L, Longley J, Lopez J, Michot JM, et al. Phase I study of the novel enhancer of zeste homolog 2 (EZH2) inhibitor GSK2816126 in patients with advanced hematologic and solid tumors. Clin Cancer Res. 2019;25:7331–9.
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360–5.
Verma SK, Tian X, La France LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of potent, selective, cellactive inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–6.
Yu T, Wang Y, Hu Q, Wu W, Wu Y, Wei W, et al. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017;8:98348–59.
Kuntz KW, Campbell JE, Keilhack H, Pollock RM, Knutson SK, Porter-Scott M, et al. The importance of being me: magic methyls, methyltransferase inhibitors, and the discovery of tazemetostat. J Med Chem. 2016;59:1556–64.
Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7.
FDA Approves Tazemetostat for Epithelioid Sarcoma. https://ascopost.com/issues/february-10-2020/fda-approves-tazemetostat-for-epithelioid-sarcoma/. Accessed 9 Dec 2020.
Morschhauser F, Salles G , McKay P, Tilly H, Schmitt A, Gerecitano J. et al. Interim report from a phase 2 multicenter study of tazemetostat, an Ezh2 inhibitor, in patients with R/R B‐cell non‐hodgkin lymphomas. https://onlinelibrary.wiley.com/doi/full/https://doi.org/10.1002/hon.2437_3. Accessed 9 Dec 2020.
Zauderer MG, Szlosarek P, Moulec SL, Popat S, Taylor P, Planchard D, et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with R/R (R/R) MM (MM) with BAP1 inactivation. J Clin Oncol. 2018;36:8515–8515.
Italiano A, Soria JC, Toulmonde M, Michot JM, Carlo Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in R/R B-cell NHLand advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–59.
Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, et al. EPZ011989, a potent, orally available EZH2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6:491–5.
Gehling VS, Vaswani RG, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, et al. Discovery, design, and synthesis of indole-based EZH2 inhibitors. Bioorg Med Chem Lett. 2015;25:3644–9.
Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, et al. Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J Med Chem. 2016;59:9928–41.
Our programs. https://www.constellationpharma.com/our-programs/. Accessed on 9 Sep 2020.
Harb W, Abramson J, Lunning M, Goy A, Maddocks K, Lebedinsky C, et al. A phase 1 study of CPI-1205, a small molecule inhibitor of EZH2, in patients with B-cell lymphomas. Ann Oncol. 2018;29:III7.
Song X, Gao T, Wang N, Feng Q, You X, Ye T, et al. Selective inhibition of EZH2 by ZLD1039 blocks H3K27 methylation and leads to potent anti-tumor activity in breast cancer. Sci Rep. 2016;6:20864.
Kung PP, Bingham P, Brooun A, Collins M, Deng YL, Dinh D, et al. Optimization of orally bioavailable enhancer of zeste homolog 2 (EZH2) inhibitors using ligand and propertybased design strategies: identification of development candidate (R)-5,8-dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4 dihydroisoquinolin-1(2H)-one (PF-06821497). J Med Chem. 2018;61:650–65.
Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–57.
Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108:2069–78.
Maruyama D, Tobinai K, Makita S, Ishida T, Kusumoto S, Ishitsuka K, et al. First-in-human study of the EZH1/2 dual inhibitor DS-3201b in patients with R/R non-Hodgkin lymphomas—preliminary results. Blood. 2017;130:4070.
EED Inhibitor MAK683. https://www.ncbi.nlm.nih.gov/medgen/1615803. Accessed on 9 Oct 2020.
Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol. 2017;13:381–8.
Wong M, Polly P, Liu T. The histone methyltransferase DOT1L: regulatory functions and a cancer therapy target. Am J Cancer Res. 2015;5:2823–37.
Sarno F, Nebbioso A, Altucci L. DOT1L: a key target in normal chromatin remodelling and in mixed lineage leukaemia treatment. Epigenetics. 2020;15:439–53.
Basavapathruni A, Olhava EJ, Daigle SR, Therkelsen CA, Jin L, Boriack-Sjodin PA, et al. Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm Drug Dispos. 2014;35:237–52.
Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–25.
Klaus CR, Iwanowicz D, Johnston D, Johnston D, Campbell CA, Smith JJ, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther. 2014;350:646–56.
Waters NJ, Daigle SR, Rehlaender BN, Basavapathruni A, Campbell CT, Jensen TB, et al. Exploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic target. J Control Release. 2015;220(Pt B):758–65.
Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–25.
Klaus CR, Iwanowicz D, Johnston D, Campbell CA, Smith JJ, Moyer MP, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther. 2014;350:646–56.
Waters NJ, Daigle SR, Rehlaender BN, Basavapathruni A, Campbell CT, et al. Exploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic target. J Control Release. 2015;220:758–65.
Shukla N, Wetmore C, O’Brien MM, Silverman LB, Brown P, Daigle S, et al. Final report of phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in children with R/R MLL-r acute leukemia. Blood. 2016;128:2780.
Stein EM, Garcia-Manero GG, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131:2661–9.
Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6:a026831.
Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–11.
Nepali K, Chang TY, Lai MJ, Hsu KC, Yen Y, Lin TE, Lee SB, Liou JP. Purine/purine isoster based scaffolds as new derivatives of benzamide class of HDAC inhibitors. Eur J Med Chem. 2020;196:112291.
Hassig CA, Schreiber SL. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr Opin Chem Biol. 1997;1:300–8.
Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51.
Paris M, Porcelloni M, Binaschi M, Fattori D. Histone deacetylase inhibitors: from bench to clinic. J Med Chem. 2018;51:1505–29.
Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–68.
Glauben R, Sonnenberg E, Zeitz M, Siegmund B. HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett. 2009;280:154–9.
Thaler F, Minucci S. Next generation histone deacetylase inhibitors: the answer to the search for optimized epigenetic therapies? Exp Opin Drug Disc. 2011;6:393–404.
Federico M, Bagella L. Histone deacetylase inhibitors in the treatment of hematological malignancies and solid tumors. J Biomed Biotechnol. 2011;2011:475641.
Reddy DS, Wu X, Golub VM, Dashwood WM, Dashwood RH. Measuring histone deacetylase inhibition in the brain. Curr Protoc Pharmacol. 2018;81:41.
Yoshida M, Kudo N, Kosono S, Ito A. Chemical and structural biology of protein lysine deacetylases. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:297–321.
Jiang Y, Liu J, Chen D, Yan L, Zheng W. Sirtuin inhibition: strategies, inhibitors, and therapeutic potential. Trends Pharmacol Sci. 2017;38:459–72.
Ganesan, A. Targeting the zinc-dependent histone deacetylases (HDACs) for drug discovery. In: Mai A editor. Topics in medicinal chemistry 33 Epigenetic Chemistry. Springer International Publishing; 2020.
Fournel M, Bonfils C, Hou Y, Yan T, Trachy-Bourget MC, Kalita A, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–68.
Knipstein J, Gore L. Entinostat for treatment of solid tumors and hematologic malignancies. Exp Opin Invest Drugs. 2011;20:1455–67.
Ossenkoppele GJ, Lowenberg B, Zachee P, Vey N, Breems D, Loosdrecht A, et al. A phase I first-in-human study with tefinostat—a monocyte/macrophage targeted histone deacetylase inhibitor—in patients with advanced haematological malignancies. Br J Haematol. 2013;162:191–201.
Jung DE, Park SB, Kim K, Kim C, Song SY. CG200745, an HDAC inhibitor induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the hippo path. Sci Rep. 2017;7:10921.
Vogl DT, Raje N, Sundar J, Richardson P, Hari P, Orlowski R. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for R/R multiple myeloma. Clin Cancer Res. 2017;23:3307–15.
Huang P, Almeciga-Pinto I, Jarpe M, van Duzer JH, Mazitschek R, Yang M, et al. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget. 2017;8:2694–700.
Shimizu T, LoRusso PM, Papadopoulos KP, Patnaik A, Beeram M, Smith LS. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin Cancer Res. 2014;20:5032–40.
Mehrling T, Chen Y. The alkylating-HDAC inhibition fusion principle: taking chemotherapy to the next level with the first in class molecule EDO-S101. Anticancer Agents Med Chem. 2016;16:20–8.
Ono H, Sowa Y, Horinaka M, Iizumi Y, Watanabe M, Morita M, et al. The histone deacetylase inhibitor OBP-801 and eribulin synergistically inhibit the growth of triple-negative breast cancer cells with the suppression of survivin, Bcl-XL, and the MAPK pathway. Breast Cancer Res Treat. 2018;171:43.
Kudo M, Ryoo BY, Lim HY, Kim DY, Okusaka T, Ikeda M, et al. Resminostat and sorafenib combination therapy for advanced HCCin patients previously untreated with systemic chemotherapy. J Clin Oncol. 2017;35:252–252.
Walewski J, Paszkiewicz-Kozik E, Warszewska A, Borsaru G, Moicean A, Hellmann A, et al. Final results of the phase II saphire trial of resminostat (4sc-201) in patients with R/R hodgkin lymphoma; Proceedings of the 53rd ASH Annual Meeting and Exposition; San Diego, CA, USA. 10–13 December 2011.
Ikeda M, Ohno I, Ueno H, Mitsunaga S, Hashimoto Y, Okusaka T, et al. Phase I study of resminostat, an HDAC inhibitor, combined with S-1 in patients with pre-treated biliary tract or pancreatic cancer. Invest New Drugs. 2019;37:109–17.
Tambo Y, Hosomi Y, Sakai H, Nogami N, Atagi S, Sasaki Y, et al. Phase I/II study of docetaxel combined with resminostat, an oral hydroxamic acid HDAC inhibitor, for advanced NSCLC in patients previously treated with platinum-based chemotherapy. Invest New Drugs. 2017;35:217–26.
Quintas-Cardama A, Kantarjian H, Estrov Z, Borthakur G, Cortes J, Verstovsek S. Therapy with the histone deacetylase inhibitor pracinostat for patients with MF. Leuk Res. 2012;36:1124–7.
Razak AR, Hotte SJ, Siu LL, Chen EX, Hirte HW, Powers J, et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of sb939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br J Cancer. 2011;104:756–62.
Zorzi AP, Bernstein M, Samson Y, Wall DA, Desai S, Nicksy D, et al. A phase I study of histone deacetylase inhibitor, pracinostat (sb939), in pediatric patients with refractory solid tumors: Ind203 a trial of the ncic ind program/c17 pediatric phase I consortium. Pediatr Blood Cancer. 2013;60:1868–74.
Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with jak2v617f positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150:446–55.
Finazzi G, Vannucchi AM, Martinelli V, Ruggeri M, Nobile F, Specchia G, et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol. 2013;161:688–94.
Chifotides HT, Bose P, Verstovsek S. Givinostat: an emerging treatment for polycythemia vera. Exp Opin Invest Drugs. 2020;29:525–36.
Aggarwal R, Thomas S, Pawlowska N, Bartelink I, Grabowsky J, Jahan T, et al. Inhibiting histone deacetylase as a means to reverse resistance to angiogenesis inhibitors: phase I study of Abexinostat Plus Pazopanib in advanced solid tumor malignancies. J Clin Oncol. 2017;35:1231–9.
Xynomic pharma receives fast-track designation from the US FDA for abexinostat as 4l therapy treating FL. https://xynomicpharma.com/en/xynomic-pharma-receives-fast-track-designation-from-the-us-fda-for-abexinostat-as-4l-therapy-treating-follicular-lymphoma/. Accessed 21 Oct 2020.
Sborov DW, Canella A, Hade EM, Mo X, Khountham S, Wang J, et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphoma. Leuk Lymphoma. 2017;58:2310–8.
Liva SG, Coss CC, Wang J, Blum W, Klisovic R, Bhatnagar B, Walsh K, et al. Phase I study of AR-42 and decitabine in AML. Leuk Lymphoma. 2020;61:1484–92.
Bistionastat. http://glixxlabs.com/chemical-products/bioactive-screen-leads-p6/GLXC-22064. Accessed on 21 Aug 2020.
Huang HH, Hou J, Zhang YM, Zhou YB, Jia L, Nan FJ. Phase 1 study of bisthianostat, an orally efficacious pan-HDAC inhibitor: part results of safety, pharmacokinetics and efficacy in patients with R/R multiple myeloma. Blood. 2019;134:5591.
Venugopal B, Baird R, Kristeleit RS, Plummer R, Cowan R, Stewart A, et al. Phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:4262–72.
Tjulandin S, Fedyanin M, Vladimirov VI, Kostorov V, Lisyanskaya AS, Krikunova L. A multicenter phase II study of the efficacy and safety of quisinostat (an HDAC inhibitor) in combination with paclitaxel and carboplatin chemotherapy (CT) in patients (pts) with recurrent platinum resistant high grade serous epithelial ovarian, primarily peritoneal or fallopian tube carcinoma cancer (OC). J Clin Oncol. 2017;15:5541–5541.
Child F, Ortiz-Romero PL, Alvarez R, Bagot M, Stadler R, Weichenthal M, et al. Phase II multicenter trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/sézary syndrome. Br J Dermatol. 2016;175:80–8.
Viracta announces positive phase 1b/2 data presented at oral presentation on lead program for Epstein–Barr Virus (EBV)-associated R/R lymphomas at the 2019 American Society of Hematology (ASH) Annual Meeting. https://apnews.com/press-release/prprnewswire/bf9846689be457e959b45007c79cfd70. Accessed 15 Jul 2020.
Viracta announces positive phase 1b/2 data presented at oral presentation on lead program for Epstein–Barr Virus (EBV)-associated R/R lymphomas at the 2019 American Society of Hematology (ASH) Annual Meeting. https://www.prnewswire.com/news-releases/viracta-announces-positive-phase-1b2-data-presented-at-oral-presentation-on-lead-program-for-epstein-barr-virus-ebv-associated-relapsedrefractory-lymphomas-at-the-2019-american-society-of-hematology-ash-annual-meeting-300971007.html.
Fimepinostat. https://www.curis.com/pipeline/fimepinostat/. Accessed 21 Nov 2020.
Wagner FF, Lundh M, Kaya T, McCarren P, Zhang YL, Chattopadhyay S. An isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in β-cell protection. ACS Chem Biol. 2016;11:363–74.
Yeruva SLH, Zhao F, Miller KD, Tevaarwerk AJ, Wagner LI, Gray RJ, et al. E2112: randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. NPJ Breast Cancer. 2018;4:1.
Etinostat. http://www.syndax.com/pipeline/entinostat/. Accessed 21 Jul 2020.
Grivas P, Mortazavi A, Picus J, Hahn NM, Milowsky MI, Hart LL, et al. Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes. Cancer. 2019;125:533–40.
Batlevi CL, Crump M, Andreadis C, Rizzieri D, Assouline SE, Fox S, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in R/R lymphoma. Br J Haemat. 2017;178:434–41.
Tucidinostat. https://pubchem.ncbi.nlm.nih.gov/compound/Tucidinostat. Accessed 23 Oct 2020.
Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui S, et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20:806–15.
Tresckow BV, Sayehli C, Aulitzky WE, Goebeler ME, Schwab M, Braz E, et al. Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur J Haematol. 2019;102:163–73.
CXD1. https://cellerontherapeutics.com/pipeline/. Accessed 23 Nov 2020.
Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for R/R multiple myeloma. Clin Cancer Res. 2017;23:3307–15.
Citarinostat. https://myelomaresearchnews.com/citarinostat-acy-241/. Accessed 19 Jul 2020.
Galloway TJ, Wirth LJ, Colevas AD, Gilbert J, Bauman JE, Saba NF, et al. A phase I study of CUDC-101, a multitarget inhibitor of HDACs, EGFR, and HER2, in combination with chemoradiation in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:1566–73.
Mita A, Loeffler M, Bui N, Mehrling T, Rimmel BJ, Natale RB, et al. Dose escalation of tinostamustine in patients with advanced solid tumors [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29–Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Canc. Res 2019;79(13 Suppl): Abstract nr CT023.
Garcia-Manero C, Sekeres MA, Egyed M, Breccia M, Graux C, Cavenagh JD, et al. A phase 1b/2b multicenter study of oral panobinostat plus azacitidine in adults with MDS, CMML or AML with ⩽30% blasts. Leukemia. 2017;31:2799–806.
National Medical Products Administration (NMPA) Approves Chipscreen Bioscience's Chidamide (Epidaza) for Breast Cancer Indication. https://www.prnewswire.com/news-releases/national-medical-products-administration-nmpa-approves-chipscreen-biosciences-chidamide-epidaza-for-breast-cancer-indication-300966800.html. Accessed 12 Jul 2020.
Hong J, Luesch H. Largazole: from discovery to broad-spectrum therapy. Nat Prod Rep. 2012;29:449–56.
Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–35.
Abaza YM, Kadia TM, Jabbour EJ, Konopleva MY, Borthakur G, Ferrajoli A, et al. Phase 1 dose escalation multicenter trial of pracinostat alone and in combination with azacitidine in patients with advanced hematologic malignancies. Cancer. 2017;123:4851–9.
Garcia-Manero G, Abaza Y, Takahashi K, Medeiros BC, Arellano M, Khaled SK, et al. Pracinostat plus azacitidine in older patients with newly diagnosed AML: Results of a phase 2 study. Blood Adv. 2019;3:508–18.
Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–87.
Mohammad HP, Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28:57–69.
Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer LSD1/KDM1A inhibitors in clinical trials: advances and prospects. Nat Med. 2019;25:403–18.
Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol. 2019;12:129.
Heijnen WT, De Fruyt J, Wierdsma AI, Sienaert P, Birkenhager TK. Efficacy of tranylcypromine in bipolar depression: a systematic review. J Clin Psychopharmacol. 2015;35:700–5.
Baker GB, Coutts RT, McKenna KF, Sherry-McKenna RL. Insights into the mechanisms of action of the MAO inhibitors phenelzine and tranylcypromine: a review. J Psychiatry Neurosci. 1992;17:206–14.
Ulrich S, Ricken R, Adli M. Tranylcypromine in mind (part I): review of pharmacology. Eur Neuropsychopharm. 2017;27:697–713.
Ricken R, Ulrich S, Schlattmann P, Adli M. Tranylcypromine in mind (part II): review of clinical pharmacology and meta-analysis of controlled studies in depression. Eur Neuropsychopharm. 2017;27:714–31.
Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S. Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25 Å. Biochem Biophys Res Commun. 2008;366:15–22.
Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in AML. Nat Med. 2012;18:60511.
Watts JM, Bradley TJ, Thomassen A, Dinh YA, Tejera D, et al. The lysine-specific demethylase 1 (LSD1) inhibitor tranylcypromine (TCP) in combination with ATRA is tolerable and has anti-leukemic activity in adult patients with R/R AML and MDS. Blood. 2018;132:2721.
Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent kdm1a inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33:495–511.
Somervaille T, Salamero O, Montesinos P, Willekens C, Simon JAP, Pigneux A, et al. Safety, phamacokinetics (PK), pharmacodynamics (PD) and preliminary activity in acute leukemia of Ory-1001, a first-in-class inhibitor of lysine-specific histone demethylase 1A (LSD1/KDM1A): initial results from a first-in-human phase 1 study. Blood. 2016;128:4060.
Fiskus W, Mill CP, Soldi R, Han R, Saenz DT, Nowak AJ, et al. Synergistic pre-clinical activity of targeted inhibition of KDM1A and BET proteins against human AML blast progenitor cells. Blood. 2018;132:3930.
Maes T, Mascaró C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33:495–511.
Bose P, Konopleva MY. ORY-1001: overcoming the differentiation block in AML. Cancer Cell. 2018;33:342–3.
Smitheman K, Cusan M, Liu Y, Butticello M, Pappalardi FJ, et al. Inhibition of LSD1 for the treatment of cancer. Cancer Res. 2015;75:3513–3513.
Civenni G, Zoppi G, Vazquez R, Shinde D, Paganoni A, Kokanovic A, et al. INCB059872, a novel FADdirected LSD1 Inhibitor, is active in prostate cancer models and impacts prostate cancer stem-like cells. Cancer Res. 2018;78(13 Supplement):1379–1379.
Roman VD, Ye M, Liu H, Diamond M, Chadderton A, Lo Y, et al. The evaluation of INCB059872, an FAD-directed covalent inhibitor of LSD1, in preclinical models of Ewing sarcoma. Cancer Res. 2017;77(13 Supplement):1162–1162.
Lee SH, Stubbs M, Liu XM, Diamond M, Dostalik V, Ye M, et al. Discovery of INCB059872, a novel FAD-directed LSD1 inhibitor that is effective in preclinical models of human and murine AML. Cancer Res. 2016;76:4712–4712.
Lee SH, Liu XM, Diamond M, Dostalik V, Favata M, He C, et al. The evaluation of INCB059872, an FAD-directed inhibitor of LSD1, in preclinical models of human SCLC. Cancer Res. 2016;76:Abstract nr 4704.
Ye M, Liu M, Lu J, Lo Y, Favata M, Yang G, et al. The LSD1 inhibitor INCB059872 is synergistic with ATRA in models of non-APL acute myelogenous leukemia. Cancer Res. 2016;76:4696–4696.
Jutzi J, Rienhoff H, Pahl H. Effectiveness of LSD1 inhibition for the treatment of MPN. https://library.ehaweb.org/eha/2017/22nd/181705/jonas.jutzi.effectiveness.of.lsd1.inhibition.for.the.treatment.of.mpn.html. Accessed 10 Jul 2020.
IMG-7289. https://adisinsight.springer.com/drugs/800048131. Accessed 21 Nov 2020.
Hollebecque A, de Bono J, Plummer R, Isambert N, Martin-Romano P, Baudin E, et al. Phase I study of CC-90011 in patients with advanced solid tumors and R/R non-Hodgkin lymphoma (R/R NHL). Ann Oncol. 2018;29(mdy303):002.
Hollebecque A, de Bono JS, Plummer R, Isambert N, Martin-Romano P, Baudin E, et al. Phase I study of CC-90011 in patients with advanced solid tumors and R/R non-Hodgkin lymphoma (R/R NHL). Ann Oncol. 2019;30:mdz029.
Prusevich P, Kalin JH, Ming SA, Basso M, Givens J, Li X, et al. A selective phenelzine analogue inhibitor of histone demethylase LSD1. ACS Chem Biol. 2014;9(6):1284–93.
Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SGT, Liu K, et al. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28:2155–64.
Sehrawat A, Gao L, Wang Y, Bankhead A, McWeeney SK, King CJ, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci USA. 2018;1(115):E4179–88.
Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–31.
Yu L, Wang Z, Zhang Z, Ren X, Lu X, Ding K, et al. Small molecule BET inhibitors in clinical and preclinical development and their therapeutic potential. Curr Top Med Chem. 2015;15:776–94.
Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR, et al. Drug discovery targeting bromodomain-containing protein 4(BRD4). J Med Chem. 2017;60:4533–58.
Wellaway CR, Amans D, Bamborough P, Barnett H, Bit RA, Brown JA, et al. Discovery of a bromodomain and extraterminal inhibitor with a low predicted human dose through synergistic use of encoded library technology and fragment screening. J Med Chem. 2020;63:714–46.
Stathis A, Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018;8:24–36.
Noel JK, Iwata K, Ooike S, Sugahara K, Nakamura H, Daibata M. Development of the BET bromodomain inhibitor OTX015. Mol Cancer Ther. 2013;12:C244.
Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, et al. Clin Cancer Res. 2015; 21:1628–1638.
Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin Cancer Res. 2016;22:2470–81.
Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6:17698–712.
Vázquez R, Astorgues-Xerri L, Riveiro ME, Beltrame L, Marchini S, Bertoni F, et al. OTX015 effects in triple-negative breast cancer (TNBC) models are independent of hypoxia conditions and synergistic with other anticancer agents. Cancer Res. 2015;75:3526–3526.
Civenni G, Pedrani S, Allegrini S, Bruccoleri A, Albino D, Pinton S, et al. Targeting prostate cancer stem cells (CSCs) with the novel BET bromodomain (BRD) protein inhibitor OTX015. Cancer Res. 2015;75:2625–2625.
Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara-Wilke J, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res. 2016;14:324–31.
Berenguer-Daizé C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, Bekradda M, MacKenzie S, Rezai K, Lokiec F, Riveiro M, Ouafik L. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer. 2016;139:2047–55.
Odore E, Lokiec F, Cvitkovic E, Bekradda M, Herait P, Bourdel F, et al. Phase I population pharmacokinetic assessment of the oral bromodomain inhibitor OTX015 in patients with haematologic malignancies. Clin Pharmacokinet. 2016;55:397–405.
Amorim S, Anastasios S, Gleeson M, Iyengar S, Valeria Magarotto V, Leleu X, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196-204.
Berthon C, Raffoux E, Thomas X, Vey N, Carlos Gomez-Roca C, Yee K, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–95.
Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. phase Ib trial with MK-8628/OTX015, a small molecule inhibitor ofbromodomain (BRD) and extra-terminal (BET) proteins, in patientswith selected advanced solid tumors. J Clin Oncol. 2018;36:JCO2018782292.
Albrecht BJ, Gehling VS, Hewitt MC, Vaswani RG, Côté A, Leblanc Y, Nasveschuk CG, et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J Med Chem. 2016;59(4):1330–9.
Abramson JS, Blum KA, Flinn IW, Gutierrez M, Goy A, Maris M, et al. BET inhibitor CPI-0610 is well tolerated and induces responses in diffuse large B-cell lymphoma and FL: preliminary analysis of an ongoing phase 1 study. Blood. 2015;126:1491.
Blum KA, Abramson J, Maris M, Flinn A, Goy A, Mertz J et al. A phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with R/R lymphoma. Presented at: TAT 2018 Targeted Anticancer Therapies; March 5-7, 2018; Paris, France. Abstract 410.
Kremyanskaya M, Hoffman R, Mascarenhas J, Verstovsek S, Mertz J, Garner F, et al. A phase 2 study of Cpi-0610, a bromodomain and extraterminal (BET) inhibitor, in patients with MF (MF). Blood. 2018;132:5481.
Hilton J, Cristea MC, Voskoboynik M, Postel-Vinay S, Edenfield W, Gavai A, et al. Initial results from a Phase I/IIA trial evaluating BMS-986158, an inhibitor of the bromodomain and extra-terminal (BET) proteins, in patients (pts) with advanced cancer. Ann Oncol. 2018;29(mdy279):399.
Gavai AV, Norris D, Tortolani D, O’Malley D, Zhao Y, Quesnelle CD, et al. Discovery of clinical candidate BMS-986158, an oral BET inhibitor, for the treatment of cancer. Cancer Res. 2018;78:5789–5789.
Aggarwal R, Abida W, Schweizer M, Pantuck A, Nanus D, Heath E, et al. A Phase Ib/IIa study of the BET bromodomain inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC) [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29-Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res 2019; 79: Abstract nr CT095.
The clinical candidate ZEN-3694, a BET bromodomain inhibitor, is efficacious in the treatment of a variety of solid tumor and hematological malignancies, alone or in combination with several standard of care therapies.https://www.zenithepigenetics.com/upload/media_element/36/01/2015-aacr-eortc-poster.pdf. Accessed 23 Sep 2020.
Dose escalation and expansion study of GSK525762 in combination with fulvestrant in subjects with hormone receptor-positive (HR)/human epidermal growth factor receptor 2 negative (HER2-) advanced or metastatic breast cancer. https://clinicaltrials.gov/ct2/show/NCT02964507. Accessed 15 Sep 2020.
Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 study of molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectr. 2020;4:93.
Dawson M, Stein E, Huntly B, Karadimitris A, Kamdar M, de Larrea CF, et al. A Phase I study of GSK525762, a selective bromodomain (BRD) and extra terminal protein (BET) inhibitor: results from part 1 of Phase I/II open label single agent study in patients with AML (AML). Blood. 2017;130:1377.
Dickinson M, Kamdar M, Huntly BJP, De Larrea CF, Cordoba R, Mateos MV, et al. A phase I study of molibresib (GSK525762), a selective bromodomain (BRD) and extra terminal protein (BET) inhibitor: results from part 1 of a phase I/II open label single agent study in subjects with non-Hodgkin’s lymphoma (NHL). Blood. 2018;132:1682.
Gadgeel S, Keedy V, Taylor S, Amin K, Horner T, Wu Y, et al. A first-in-human phase I dose escalation study of BET inhibitor GSK2820151 in patients with advanced or recurrent solid tumors cancers [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1–5; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2017;77(13 Suppl):Abstract nr CT104. doi:https://doi.org/10.1158/1538-7445.AM2017-CT104.
Forero-Torres A, Rosen S, Smith DC, Lesser G, Peguero J. Preliminary results from an ongoing phase 1/2 Study of INCB057643, a bromodomain and extraterminal (BET) protein inhibitor, in patients (pts) with advanced malignancies. Blood. 2017;130:4048.
ODM-207, a novel BET-bromodomain inhibitor as a therapeutic approach for the treatment of prostate and breast cancer. https://www.orion.fi/globalassets/documents/rd/congress-posters/odm-207-nonclinical-poster-2016-bjorkman-et-al.pdf. Accessed 21 Nov 2020.
Postel-Vinay S, Herbschleb K, Massard C, Woodcock V, Soria JC, Walter A, et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur J Cancer. 2019;109:103–10.
Gaudio BE, Lejeune P, Tarantelli C, Cascione L, Kwee I, et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br J Haematol. 2017;178:936–48.
Moreno V, Sepulveda JM, Vieito M, Hernández-Guerrero T, Doger B, Saavedra O, et al. Phase I study of CC-90010 in patients with advanced solid tumors and R/R NHL(R/R NHL). Ann Oncol. 2020;31:780–8.
Sing-Zan Wang J, De Vita S, Karlix JL, Cook C, Littlewood GM, Hattersley M. First-in-human study of AZD5153, a small molecule inhibitor of bromodomain protein 4 (BRD4), in patients (pts) with R/R (RR) malignant solid tumor and lymphoma: preliminary data. J Clin Oncol. 2019;37:3085–3085.
Millan DS, Alvarez Morales MA, Barr KJ, Cardillo D, Collis A, Dinsmore CJ, et al. FT-1101: a structurally distinct pan-BET bromodomain inhibitor with activity in preclinical models of hematologic malignancies. Blood. 2015;126:1367.
Patel MR, Garcia-Manero G, Paquette R, Dinner S, Donnellan WB, Grunwald MR, et al. Phase 1 dose escalation and expansion study to determine safety, tolerability, pharmacokinetics, and pharmacodynamics of the BET inhibitor FT-1101 as a single agent in patients with R/R hematologic malignancies. Blood. 2019;134:3907.
Patnaik A, Carvajal RD, Komatsubara KM, Britten CD, Wesolowski R, Michelson G, et al. Phase Ib/2a study of PLX51107, a small molecule BET inhibitor, in subjects with advanced hematological malignancies and solid tumors. J Clin Oncol. 2018;36:2550–2550.
Mahadevan D, Chiorean EG, Harris WB, Von Hoff D, Stejskal-Barnett A, Qi W, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer. 2012;48:3319–27.
Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, et al. PROTACs: great opportunities for academia and industry. Signal Transduct Target Therapy. 2019;4:64.
Troup IR, Fallan C, Baud MGJ. Current strategies for the design of PROTAC linkers: a critical review. Explor Target Antitumor Therapy. 2020;1:273–312.
PROTAC linkers. https://www.creative-biolabs.com/protac/linker-design-andoptimization.html. Accessed 20 Aug 2020.
Tissue expression of frequently recruited E3 ligase in PROTAC design. https://www.proteinatlas.org/ENSG00000134086-VHL/tissue. Accesesed 21 Sep 2020.
Liang Y, Nandakumar KS, Cheng K, et al. Design and pharmaceutical applications of proteolysis-targeting chimeric molecules. Biochem Pharmacol. 2020;182:114211.
Wu H, Yang K, Zhang Z, Leisten ED, Li Z, Xie H. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem. 2019;62:7042–57.
An Z, Lv W, Su S, Wu W, Rao Y, et al. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell. 2019;10:606–9.
Yang K, Wu H, Zhongrui Z, Leisten ED, Nie X, Liu B, et al. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting Von Hippel-Lindau (VHL) E3 ubiquitin ligase. ACS Med Chem Lett. 2020;11:575–81.
Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang CY, Liu L, et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J Med Chem. 2018;61:6685–704.
Raina K, Lu J, Qian Y, Altieri M, Gordon D, Ann Marie K, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2016;113:7124–9.
Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–81.
Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77:2476–87.
Zopp V, Hughes SJ, Maniaci C, Testa A, Wieshofer TGC, ManfKoegl M, et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem. 2019;62:699–726.
Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew Chem Int Ed. 2017;56:5738–43.
Wayne N, Mishra P, Bolon DN. Hsp90 and client protein maturation. Methods Mol Biol. 2011;787:33–44.
Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72.
Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation bybenzoquinone ansamycins: essential role for stress proteins in oncogenictransformation. Proc Natl Acad Sci U S A. 1994;91:8324–8.
Kim SH, Kang JG, Kim CS, Ihm SH, Choi MG, Yoo HJ, et al. The heat shock protein 90 inhibitor SNX5422 has a synergistic activity with histone deacetylase inhibitors in induction of death of anaplastic thyroid carcinoma cells. Endocrine. 2016;51:274–82.
Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, et al. HDAC6 inhibition enhances 17-AA gemediated abrogation of hsp90 chaperone function in human leukemia cells. Blood. 2008;112:1886–93.
Rahmani M, Yu C, Dai Y, Reese E, Ahmed W, Dent P, et al. Coadministration of the heat shock protein 90 antagonist 17-allylamino-17-demethoxygeldanamycin with suberoylanilide hydroxamic acid or sodium butyrate synergistically induces apoptosis in human leukemia cells. Cancer Res. 2003;63:8420–7.
George P, Bali P, Annavarapu S, Scuto A, Fiskus W, Guo F, et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–76.
Deskin B, Shan B, Lasky J. Targeting the Hdac6/hsp90 axis to destabilize the Notch1 receptor in NSCLC. Am J Respir Crit Care Med. 2016;193:A2374.
Ojha R, Nepali K, Chen CH, Chuang KH, Wu TY, Lin TE, et al. Isoindoline scaffold-based dual inhibitors of HDAC6 and HSP90 suppressing the growth of lung cancer in vitro and in vivo. Eur J Med Chem. 2020;190:112086.
Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RWC, et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell. 2017;171:1284-1300 e21.
Pathania R, Ramachandran S, Mariappan G, Thakur P, Shi H, Choi JH, et al. Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res. 2016;76:3224–35.
Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced NSCLC. Cancer Discov. 2011;1:598–607.
Yang XW, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE, et al. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61:7025–9.
Ecke I, Petry F, Rosenberger A, Tauber S, Monkemeyer S, Hess I, et al. Cancer Res. 2009;69:887–95.
Yuan Z, Chen S, Gao C, Dai Q, Zhang CL, Sun Q, et al. Development of a versatile DNMT and HDAC inhibitor C02S modulating multiple cancer hallmarks for breast cancer therapy. Cancer Cell. 2015;28:57–69.
Laugesen A, Helin K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell. 2014;14:735–51.
Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28:57–69.
Kalin JH, Wu M, Gomez AV, Song Y, Das Y, Hayward D, et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun. 2018;9:53.
Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sánchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–71.
Zhang X, Zegar T, Weisew T, Hamdan FH, Berger BT, Lucas R. Characterization of a dual BET/HDAC inhibitor for treatment of pancreatic ductal adenocarcinoma. Int J Cancer. 2020;147:2847–61.
Grinshtein N, Rioseco CC, Marcellus R, Uehling D, Aman A, Lun X, et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget. 2016;13:59360–76.
Yamaguchi J, Sasaki M, Sato Y, Itatsu K, Harada K, Zen Y, et al. Histone deacetylase inhibitor (SAHA) and repression of EZH2 synergistically inhibit proliferation of gallbladder carcinoma. Cancer Sci. 2010;101:355–62.
Lue JK, Prabhu SA, Liu Y, Gonzalez Y, Verma A, Mundi PS, et al. Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin Cancer Res. 2019;25:5271–83.
Huang JP, Ling K. EZH2 and histone deacetylase inhibitors induce apoptosis in TNBC cells by differentially increasing H3 Lys27 acetylation in the BIM gene promoter and enhancers. Oncol Lett. 2017;14:5735–42.
Takashina T, Kinoshita I, Kikuchi J, Shimizu Y, Sakakibara-Konishi J, Oizumi S, et al. Combined inhibition of EZH2 and histone deacetylases as a potential epigenetic therapy for non-small-cell lung cancer cells. Cancer Sci. 2016;107:955–62.
Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34.
Romanelli A, Stazi G, Fioravanti R, Zwergel C, Di Bello E, Perrone PS, et al. Design of first-in-class dual EZH2/HDAC inhibitor: biochemical activity and biological evaluation in cancer cells. ACS Med Chem Lett. 2020;11:977–83.
Thakur A, Tawa GJ, Henderson MJ, Danchik C, Liu S, Shah P, et al. Design, synthesis, and biological evaluation of quinazolin-4-one-based hydroxamic acids as dual PI3K/HDAC inhibitors. J Med Chem. 2020;63:4256–92.
Chao MW, Lai MJ, Liou JP, Chang YL, Wang JC, Pan SL, et al. The synergic effect of vincristine and vorinostat in leukemia in vitro and in vivo. J Hematol Oncol. 2015;8:82.
Lamaa D, Lin HP, Zig L, Bauvais C, Bollot G, Bignon J, et al. Design and synthesis of tubulin and histone deacetylase inhibitor based on iso combretastatin A-4. J Med Chem. 2018;61:6574–91.
Nepali K, Sharma S, Sharma M, Bedi PMS, Dhar KL. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur J Med Chem. 2014;77:422–87.
Piekarz R, Bates S. A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr Pharm Des. 2004;10:2289–98.
Richardson PG, Harvey RD, Laubach JP, Moreau P, Lonial S, San-Miguel JF. Panobinostat for the treatment of relapsed or R/R multiple myeloma: pharmacology and clinical outcomes. Exp Rev Clin Pharmacol. 2016;9:35–48.
Srinivas NR. Clinical pharmacokinetics of panobinostat, a novel histone deacetylase (HDAC) inhibitor: review and perspectives. Xenobiotica. 2017;47:354–68.
Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107:600–8.
Nepali K, Chang TY, Lai MJ, Hsu KC, Yen Y, Lin TE, et al. Purine/purine isoster based scaffolds as new derivatives of benzamide class of HDAC inhibitors. Eur J Med Chem. 2020;196:112291.
Lee HY, Nepali K, Huang FI, Chang CY, Lai MJ, Li YH, et al. (N Hydroxycarbonylbenylamino) quinolines as selective histone deacetylase 6 inhibitors suppress growth of multiple myeloma in vitro and in vivo. J Med Chem. 2018;61:905–17.
Lee HY, Tsai AC, Chen MC, Shen PJ, Cheng YC, Kuo CC, Pan SL, Liu YM, et al. Azaindolyl sulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells. J Med Chem. 2014;57:4009–22.
Loredana S, Hideshima T, Kung AL, Tseng JC, David T, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–89.
Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. PNAS. 2003;100:4389–94.
Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, Tubastatin A. J Am Chem Soc. 2010;132:10842–6.
Kang SP, Ramirez J, House L, Zhang W, Mirkov S, Liu W, et al. A pharmacogenetic study of vorinostat glucuronidation. Pharmacogenet Genomics. 2010;20:638.
Wong NS, Seah EZ, Wang LZ, Yeo WL, Yap HL, Chuah B, et al. Impact of UDP-gluconoryltransferase 2B17 genotype on vorinostat metabolism and clinical outcomes in Asian women with breast cancer. Pharmacogenet Genomics. 2011;21:760–8.
Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–9.
Assem ESK, Peh KH, Wan BY, Middleton BJ, Dines J, Marson CM, et al. Effects of a selection of histone deacetylase inhibitors on mast cell activation and airway and colonic smooth muscle contraction. Int Immunopharmacol. 2008;8:1793–801.
Bresciani A, Ontoria JM, Biancofiore I, Cellucci A, Ciammaichella A, Di Marco A, et al. Improved selective class I HDAC and novel selective HDAC3 inhibitors: beyond hydroxamic acids and benzamides. ACS Med Chem Lett. 2018;10:481–6.
Allen FH. The Cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;58:380–8.
Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9:319–25.
Vianello P, Botrugno OA, Cappa A, Zuffo RD, Dessanti P, Mai A. Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J Med Chem. 2016;59:1501–17.
Rotili D, Tomassi S, Conte M, Benedetti R, Tortorici M, Ciossani G, et al. Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities. J Med Chem. 2014;57:42–55.
Gehling VS, McGrath JP, Duplessis M, Khanna A, Brucelle F, Vaswani RG, et al. Design and synthesis of styrenylcyclopropylamine LSD1 inhibitors. ACS Med Chem Lett. 2020;11:1213–20.
Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7.
Diamantis N, Banerji U. Antibody drug conjugates an emerging class of cancer treatment. Br J Cancer. 2016;114:362–7.
Sievers EL, Senter PD. Antibodyedrug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29.
Cini E, Faltoni V, Petricci E, Taddei M, Salvini L, Giannini G, et al. Antibody drug conjugates (ADCs) charged with HDAC inhibitor for targeted epigenetic modulation. Chem Sci. 2018;9:6490–6.
Milazzo FM, Vesci L, Anastasi AM, Chiapparino C, Rosi A, Giuseppe G, et al. ErbB2 targeted epigenetic modulation: anti-tumor efficacy of the ADC trastuzumab-HDACi ST8176AA1. Front Oncol. 2020;9:1534.
Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D, Raya A. CRISPR/Cas9-mediated genome editing of epigenetic factors for cancer therapy. Cell Stem Cell. 2017;21:431–47.
Gersbach CA. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143–9.
Liao HK, Hatanaka F, Araoka T, Reddy P, Wu MZ, Sui Y, et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell. 2017;171:1495–507.
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer E, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12:326–8.
Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34:1060–5.
Acknowledgements
Jing Ping Liou is supported by the Ministry of Science and Technology, Taiwan (Grant No. MOST 109-2113-M-038-001). Kunal Nepali is supported by grants from Ministry of Science and Technology, Taiwan [Grant No. MOST108-2320-B-038-010-MY2 (2-1)] and Taipei Medical University, Taiwan (Grant No. TMU107-AE1-B33). NC, NUT carcinoma.
Funding
This work received no external funding.
Author information
Authors and Affiliations
Contributions
Drafting and literature analysis: KN; conceptualization, guidance, and supervision: JPL. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Kunal Nepali and Jing Ping Liou are employees of Taipei Medical Univeristy.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nepali, K., Liou, JP. Recent developments in epigenetic cancer therapeutics: clinical advancement and emerging trends. J Biomed Sci 28, 27 (2021). https://doi.org/10.1186/s12929-021-00721-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-021-00721-x