
Abstract
Background
Whole-exome sequencing (WES) significantly improves the diagnosis of the etiology of fetal structural anomalies. This study aims to evaluate the diagnostic value of prenatal WES and to investigate the pathogenic variants in structurally abnormal fetuses.
Methods
We recruited 144 fetuses with structural anomalies between 14 and 2020 and 15 December 2021 in the study. Genetic screening was performed by WES combined with karyotyping and chromosomal microarray analysis. The molecular diagnostic yield of prenatal WES for each type of fetal structural anomaly and the identified pathogenic genes and mutations were reported.
Results
In this study, we retrospectively analyzed the clinical and genetic data of 145 structurally anomalous fetuses. These cases were classified into 9 phenotypic classes based on antenatal ultrasound findings. Thirty-eight pathogenic variants in 24 genes were identified in 35 of the 145 cases, including 14 novel variants in 13 genes (EP300, MYH3, TSC2, MMP9, CPLANE1, INVS, COL1A1, EYA1, TTC21B, MKS1, COL11A2, PDHA1 and L1CAM). Five additional pathogenic variants were classified as incidental findings. Our study showed that the overall diagnosis rate of WES was 28.1% (27/96) in the parent-fetus trio cases and 16.3% (8/49) in the proband-only cases. Fetuses with musculoskeletal anomalies had the highest diagnostic yield (51.4%, 19/37). In addition, FGFR3 and COL1A1 were the most common pathogenic genes.
Conclusions
Our work expands the mutation spectrum of the genes associated with fetal structural anomalies and provides valuable information for future parental genetic counselling and pregnancy management of the structurally anomalous fetuses.
Similar content being viewed by others


Background
Prenatal ultrasound is a routine screening method for fetal malformations, identifying fetal structural anomalies in approximately 3% of fetuses [1]. The anomalies seen in different tissues and organs of the body are commonly associated with a variety of diseases, including heart defects, skeletal dysplasia, congenital anomalies of the kidneys and urinary tracts (CAKUT), craniofacial anomalies, and nervous system abnormalities. However, due to the limitations of prenatal ultrasonography, it is often difficult to fully determine the fetal phenotype, making the diagnosis of fetal structural anomalies more challenging.
Fetal structural anomalies are usually caused by pathogenic sequence variants and chromosomal aberrations (such as chromosomal aneuploidies and copy number variants) in development-related genes [2]. Soft ultrasound markers, such as increased nuchal translucency, thickened nuchal skin fold, amniotic fluid abnormalities, and single umbilical artery, may also indicate a risk for genetic disease [3]. In prenatal genetic testing, sequence variants are often detected by whole-exome sequencing (WES), while chromosomal aberrations are detected by karyotyping and chromosomal microarray analysis (CMA). Accurate genetic diagnosis is essential not only for the obstetricians to assess the prognosis of fetuses with ultrasound anomalies, but also for the parents to make an informed decisions about pregnancy. It is also helpful in the planning of neonatal management. However, due to the high degree of clinical and genetic heterogeneity and the fact that the vast majority of cases are sporadic, it is extremely challenging to determine the disease-causing variants responsible for the fetal structural anomalies.
WES has become a powerful and cost-effective diagnostic approach for genetic disorders [4, 5]. Several studies have reported the successful application of WES in the prenatal genetic diagnosis of fetal structural anomalies in combination with karyotyping and CMA [6,7,8,9,10,11,12,13,14]. The diagnostic yield of prenatal WES ranged from 8.5 to 39.4% in different studies [15,16,17,18,19]. Many disease-causing genes, such as LBR, SLC26A2, FLNB, COL1A2, COL2A1, and FGFR3, have been found by WES to be frequently associated with skeletal dysplasia in fetuses. Nevertheless, incomplete phenotypic identification due to the limitations of ultrasound examination and insufficient data on the functions of many candidate genes make it very difficult to determine the clinical significance of variants identified by WES [20,21,22]. The underlying genetic causes of more than 60% of the cases with fetal structural anomalies are still unknown. More phenotypic and genotypic data will improve the accuracy of genetic interpretation and increase our understanding of the pathogenesis of fetal structural anomalies.
In this study, we retrospectively analyzed the clinical and genetic data of 145 structurally anomalous fetuses. These cases were classified into 9 phenotypic classes based on antenatal ultrasound findings. Genetic screening was performed by WES combined with karyotyping and CMA. The molecular diagnostic yield of prenatal WES for each type of fetal structural anomaly and the identified pathogenic genes and mutations were reported. These data provide valuable information for advancing the clinical application of prenatal WES and for establishing systemic genotype-phenotype associations in prenatal diagnosis.
Methods
Participants recruitment and DNA preparation
From 14 to 2020 to 15 December 2021, we recruited 145 pregnancies in which the fetuses showed structural anomalies during 11–32 weeks by prenatal ultrasonography at the medical genetics centre of the Maternal and Child Health Hospital (Hubei, China). The fetuses had all undergone common tests such as intrauterine infection and autoimmune screening at the hospital by obstetricians. Only those fetuses with no clear cause underwent further genetic screening. Clinical information including maternal age, gestational age, family history, and obstetric history were recorded in detail. This study was approved by the institutional ethics committee. All parents of the fetuses agreed to participate in the study and provided signed informed consent. Clinical geneticists were mainly responsible for interviewing participants, collecting data/samples and providing pre- and post-test genetic counselling to participants. Human phenotype ontology (HPO) terms were used to standardize clinical features in the study.
Sample extraction was performed at different stages depending on the situation. For most pregnant women, amniocentesis was performed at 18–32 weeks of gestation to collect amniotic fluid. A few pregnant women underwent chorionic villus sampling at 12–14 weeks of gestation. Genomic DNA was extracted from amniotic fluid/ chorionic villi, and peripheral blood samples from both biological parents using the Qiagen DNA Blood Midi/ Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol.
Whole-exome sequencing and bioinformatics analysis
WES of parent-fetus trios (fetuses and parents) and proband-only (fetuses) was performed on DNA samples at Berry Genomics Corporation. WES samples are also subjected to short tandem repeats (STR) to exclude maternal contamination for parentage analysis. Massively parallel sequencing was performed using NanoWES (Berry Genomics, China) and the Novaseq6000 platform (Illumina, San Diego, USA). The following metrics were achieved for all samples: mean coverage depth was ~ 100×, and ~ 95% of target bases (exons and ± 20 intronic nucleotides flanking the exon-intron boundaries of all nuclear genes) were interrogated at > 20× read depth.
Raw image files were processed using CASAVA v1.82 for base calling to generate raw data. The raw sequence data were aligned to the human reference genome (hg38) using the Burrows-Wheeler Aligner tool. The PCR duplicates were removed using Picard v1.57 (http://picard.sourceforge.net/). Single nucleotide variants (SNV) and insertion/deletion (InDel) variants were detected using the Verita Trekker® Variants Detection System and GATK (https://software.broadinstitute.org/gatk/). Detected variants were annotated using ANNOVAR (http://annovar.openbioinformatics.org/en/latest/) and Enliven® Variants Annotation Interpretation System, authorized by Berry Genomics. Nonsynonymous, splicing, and InDel variants were filtered by the 1000 Genome Project (http://browser.1000genomes.org), gnomAD (http://gnomad.broadinstitute.org/), Berrybig data population database, dbSNP (http://www.ncbi.nlm.nih.gov/snp) etc. Variants with a minor allele frequency (MAF) > 0.01 were discarded. The whole exome was analyzed and late onset disorders were filtered out for each case in this study.
Variant interpretation and reporting
Relevant inheritance patterns, family history and clinical manifestations were considered during variant interpretation. Variants relevant to the corresponding phenotypes were evaluated for pathogenicity according to the adapted American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). Genetic variants were screened under the combined consideration of pathogenicity prediction using multiple tools (REVEL, SIFT, Polyphen2, FATHMM, CADD, Mutation Assessor, SPIDEX, etc.), relative hits from different databases (OMIM, http://www.omim.org; HGMD, http://www.hgmd.org; ClinVar, http://www.ncbi.nlm.nih.gov/clinvar; and HPO, https://hpo.jax.org/app/) and scientific reports. Sanger sequencing was used to confirm the inheritance of the likely pathogenic/ pathogenic variants for parent-fetus trios and proband-only cases. CNV-calling was performed using Sprinkle, a comprehensive tool developed by Berry Genetics. More specifically, XHMM PCA was used to remove sequencing background. CNVKit fixed module was used for GC and bias correction, copy number calculation in exons and long fragment regions, and CNV identification. The window size was 200 bp and the normal reference included 30 samples. Interpretation was based on CNV type, size, frequency, number of key genes and recurrence region according to ACMG guidelines. DECIPHER (https://www.deciphergenomics.org/), DGV (http://dgv.tcag.ca/dgv/app/home) and ClinGen (https://dosage.clinicalgenome.org/) were used to assess CNV pathogenicity. Copy number variants (CNVs) identified by WES were further investigated by quantitative PCR.
To report the genetic diagnostic result for each fetus, clinical phenotype-related and genetic pattern compatible variants with sufficient evidence of pathogenicity were included in the main report. Variants related to the prenatal indications but with insufficient evidence of inheritance pattern and/or pathogenicity were included in the secondary report. Variants of uncertain significance (VUS) were occasionally included when there was a strong indication for reporting (e.g., the VUS was compound heterozygous with a pathogenic variant). The highly penetrant pathogenic variants that were unrelated to the fetal phenotypes were classified as incidental findings. These variants were predicted to cause moderate to severe childhood onset disorders, such as neurodevelopmental disorders and metabolic disorders. Additional relevant tests were usually carried out on the fetuses before or after birth. The results are explained in detail to the parents by clinical geneticists. Statistical comparisons were made using the Chi-square test.
Results
Sample characteristics
The 145 cases were classified into 9 phenotypic groups based on the ultrasound results (Table S1). Most cases showed abnormalities in the musculoskeletal system (n = 37), multisystem (n = 36), genitourinary system (n = 24), and nervous system (n = 13) (Table 1). Other common soft ultrasound markers including increased nuchal translucency, thickened nuchal skin fold, amniotic fluid abnormalities, and single umbilical artery, were categorized into miscellaneous abnormalities of the prenatal birth and development group (n = 12). The 145 cases include 96 fetus-parental trios and 49 proband-only cases. In all cases, the chromosomal aberrations were excluded by means of karyotyping and CMA.
WES molecular diagnostic rate
There were 35 cases identified by WES with likely pathogenic/pathogenic variants closely associated with the abnormal fetal phenotypes, resulting in an overall diagnostic rate of 24.1% (35/145). Four cases (2.8%, 4/145) carrying 5 likely pathogenic/pathogenic variants that were not considered phenotypically correlated were classified as incidental findings. In the parent-fetus trio cases (n = 96), the diagnostic rate of WES was 28.1% (27/96). In contrast, the diagnostic rate of WES in the proband-only cases (n = 49) was 16.3% (8/49) (Table 1 and and Supplementary file 1). The diagnostic rate of the parent-fetus trio group was significantly higher than that of the proband-only group (p < 0.05). In addition, variants were classified as VUSs in 13 cases (Table S2). The proportion of VUSs in the proband-only samples (14.3%, 7/49) was higher than that of the fetus-parental trios (6.25%, 6/96). The consequences of these variants have not yet been determined.
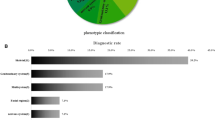
The diagnostic yield varied between the phenotypic groups (Fig. 1A), with the highest diagnostic yield in the musculoskeletal system anomalies (51.4%, 19/37), followed by multisystem anomalies (22.2%, 8/36), other prenatal anomalies (16.7%, 2/12), genitourinary system anomalies (12.5%, 3/24) and nervous system anomalies (15.4%, 2/13). A low yield of genetic diagnosis was observed in the group of craniofacial anomalies (8.3%, 1/12).

Characteristics of the potential diagnoses in fetuses with structural abnormalities. A Proportion of diagnostic genetic variants identified in fetuses with each phenotypic abnormality. B Potential diagnostic gene and gene frequency detected by WES
Mutation type and inheritance pattern of the pathogenic variants
A total of 38 different pathogenic variants in 24 genes were identified in 35 of the 145 cases, including 8 frameshifts, 2 stopgains, 5 splicings, 2 in-frame deletions, 19 nonsynonymous changes, 1 start_lost and 1 CNV (Table 2). Compound heterozygous variants were detected in 7 cases (Table 2). Of the 35 cases, 68.6% (n = 24) were autosomal dominant (AD), 22.8% (n = 8) were autosomal recessive (AR), and 8.6% (n = 3) were X-linked (XL) (Table 3). Most AD cases were associated with de novo variants (75%, n = 18/24; Table 3), whereas most AR cases were associated with compound heterozygous variants (87.5%, n = 7/8; Table 3).
Novel variants identified in the definitive diagnostic cases
Importantly, 14 novel pathogenic variants were identified in the study (Table 2). Five novel pathogenic/likely pathogenic variants, including the c.151 C > T and c.929del in MMP9, c.3977T > G in COL1A1, c.3130_3131dup in TTC21B, and c.3850 C > T in COL11A2 were found in 4 cases with skeletal dysplasia. Six novel pathogenic/likely pathogenic variants, including c.3247_3248 + 1del in MYH3, c.5239_5256del in TSC2, c.2854_2855insCT in CPLANE1, c.805_806del in INVS, c.1698 + 2T > C in EYA1 and c.1407 + 66G > A in MKS1, were found in six cases with multiple abnormalities. Two novel variants in L1CAM (c.3130_3131dup) and PDHA1 (c.766G > A) were detected in two cases with central nervous system abnormalities. A pathogenic variant, c.5527dup in EP300, was identified in the case with craniofacial abnormalities.
Representative diagnostic cases using prenatal WES
A pathogenic splicing variant in SMPD4 in trans with a deletion of 59.72 kb (chr2: 130,142,742–130,202,459) spanning SMPD4 was found in a fetus with arthrogryposis (case 2014, Table 2) using WES-based CNV analysis. SMPD4 is the gene responsible for a developmental disorder characterized by microcephaly and congenital arthrogryposis [23], which was consistently observed in the aforementioned case.
Case 2997
carried a homozygous stopgain variant in BBS2 inherited from his parents. This mutation has been previously reported in patients affected with Bardet-Biedl syndrome 2. In addition to renal dysplasia, BBS2 mutations often cause retinitis pigmentosa, a progressive retinal degenerative disease that cannot be observed in the prenatal period. According to the family history, the proband’s brother has mild intellectual disability and night blindness (as early symptoms of retinitis pigmentosa). The clinical significance of the BBS2 variant was further determined by the family history.
Pathogenic variants in FGFR3 are known to cause autosomal dominant achondroplasia and hypochondroplasia. However, a homozygous missense variant in FGFR3 was identified in one case (case 3084) with shortened long bones (Table 2). Further pedigree analysis revealed that the homozygous FGFR3 variant (c.1138G > A, p.G380R) was inherited from the proband’s parents affected by dwarfism. Two pathogenic gene variants (FGFR3, SOS1) were detected by WES in a fetus (case 4714) with shortened long bones, multiple kidneys and polyhydramnios by WES. These two diagnoses could clearly explain the structural abnormalities of the fetus.
Incidental findings
Incidental genetic variations were detected in four cases (Table 4). These variants were predicted to cause conditions unrelated or partially related to the fetal phenotypes. For example, in case 2092 with skin oedema, pleural effusion, and talipes equinovarus, two pathogenic compound heterozygous variants in the TTN gene were identified. TTN is usually associated with cardiomyopathy and muscular dystrophy, which was not relevant to the ultrasound findings in this case. Another example is a de novo heterozygous variant (c.1019 C > T, p.T340M) in FGFR1, which was identified in case 8212 with abnormalities of the cerebral ventricles and septum pellucidum and the agenesis of the corpus callosum. However, FGFR1 mutations are often associated with Hartsfield syndrome, which is characterized by holoprosencephaly, ectrodactyly and cleft lip/palate [24].
A de novo heterozygous variant in the TRIP12 gene was detected in the case 2907 with abnormal feet. Pathogenic variants in TRIP12 are often associated with neurodevelopmental disorders, such as developmental delay and intellectual disability. The clinical significance of TRIP12 variants should be confirmed by adequate postnatal follow-up. ARID1B pathogenic variants are associated with several clinical features, including intellectual disability, developmental delay, severe speech delay, corpus callosum abnormalities, etc. The ARID1B variant was classified as an incidental finding in case 6428, because most of the clinical features associated with neurodevelopmental disorders are difficult to observe on prenatal ultrasound.
Genes that frequently associated with fetal structural abnormalities
Variants in FGFR3 (n = 9) and COL1A1 (n = 4) were the most common diagnostic findings in this study (Fig. 1B). These variants were all found to be responsible for the phenotypes observed in the fetuses. Four pathogenic missense variants in FGFR3 were found in 9 cases (case 3084, 2079, 6411, 5588, 3078, 3031, 2919, 4714, 4743) with skeletal dysplasia. The variants c.1138G > A in FGFR3 were detected six times. Four missense variants in COL1A1 were detected in four different cases (case 2051, 6429, 1768 and 0543, Table 2) with shortened long bones or osteogenesis hypoplasia.
To summarize the pathogenic genes associated with fetal structural abnormalities, we performed a comparative analysis of our results with other 8 recently reported cohort studies that included at least 90 cases of fetal structural abnormalities [13,14,15,16,17,18,19, 25]. Among the pathogenic genes identified in our cohort, 12 genes were found to be novel, while 12 genes had already been identified in other studies (Fig. 2). The most frequently identified genes were FGFR3 and COL1A1, which were shared by 7 studies. The genes TSC2 and SOS1 were reported in five studies, DYNC2H1, MYH3 and KMT2D were detected in four cohort studies. COL2A1, TMEM67, BBS2, FGFR2, L1CAM, FLNA, FLNB, ANKRD11, TFAP2A, C5orf42, etc. were found in studies. Details of the disease-causing genes are shown in Fig. 2.

Genes associated with fetal structural abnormalities that have been identified in studies. The horizontal column (blue) represents the number of reported pathogenic genes in each study. The vertical column (black) represents the number of reported pathogenic genes in each intersection. An intersection means a group of pathogenic genes that was shared by one, two, or more studies. The black dots linked by black lines indicate the information that pathogenic genes in each intersection are shared by which studies. The gene symbols in some intersections are labeled
Discussion
This study reported a retrospective analysis of the clinical and genetic data from 145 structurally anomalous fetuses. A total of 38 pathogenic variants in 24 genes were identified in 35 of the 145 cases, including 14 novel variants in 13 genes. The diagnostic rates for the parent-fetus trios and the proband-only samples were 28.1% (27/96) and 16.3% (8/49), respectively. These findings provide valuable information to promote the clinical application of prenatal WES and to establish genotype-phenotype associations in prenatal diagnosis.
Several recent studies have shown that prenatal WES achieves diagnostic rates of 20–24% in fetuses with structural anomalies with normal karyotype and CMA [16, 26, 27]. In this retrospective study, we found a diagnostic yield of 24.1% (35/145) by WES, which is consistent with the rates reported in studies excluding chromosomal abnormalities. The different diagnostic rates could be affected by many factors, such as cohort size, variant interpretation criteria, diversity of anomalies and inclusion criteria [22]. A molecular diagnosis rate of 28.1% (27/96) was achieved in the fetus-parental trio samples. However, the diagnosis rate was decreased to 16.3% (8/49) in proband-only samples. This indicated that WES with trios would have a higher detection sensitivity than proband-only samples, which is consistent with previous studies [16]. It is likely that trio-WES is more effective in detecting the de novo and compound heterozygous variants. Among the 35 cases with a specific molecular diagnosis in this study, the pathogenic variants in 20 cases were de novo, accounting for 57.1%. This result suggests that de novo variants may be the predominant mutation in fetuses with structural anomalies. In addition, 15 fetuses had pathogenic variants inherited from their parents, suggesting the importance of genetic counselling and carrier screening for pregnant women.
The diagnostic yield of 51.4% gene variants in cases with musculoskeletal anomalies was significantly higher than in other cases with multisystem anomalies (22.2%), nervous system anomalies (15.1%), and genitourinary system anomalies (12.5%). Furthermore, we found a higher diagnostic yield in fetuses with musculoskeletal anomalies (19/37, 51.4%) than in those with an anomaly excluding the skeletal anomalies (8/72, 11.1%). This may be because the fetuses with abnormal skeletal system development have more distinct phenotypic features, just as other researchers have claimed that a specific organ system anomaly may have a higher diagnostic yield than a single structural anomaly [21].
FGFR3 is a pathogenic gene that causes for achondroplasia with shortened long bones and over 80% of cases have been found with spontaneous gene mutations [28]. Four heterozygous variants in FGFR3 were identified in 9 cases of skeletal dysplasia. Among these, a homozygous FGFR3 variant (G380R) in case 3084 was inherited from the parents of the proband affected by dwarfism, which is a rare finding. Six cases carried the same pathogenic variant G380R in FGFR3. Of these, two cases carrying the G380R variant (3084, 3078) presented with shortened long bones, and four other FGFR3 cases (2079, 5588, 4714, 4743) presented with shortened long bones and polyhydramnios. Case 6411, carrying the N540K variant, presented with short femur length only. However, the remaining two cases (3031 and 2919) with thanatophoric dysplasia had short limbs and a small chest. In addition, the FGFR3 G380R variant was found in the father with dwarfism, but not in the fetus with skeletal dysplasia, indicating the added value of trio WES.
On the other hand, WES can detect incidental findings that cannot be detected by prenatal imaging. For example, neurodevelopmental disorders such as developmental delay and intellectual disability cannot be detected by routine prenatal testing. Case 2907 with abnormal feet was found to carry a de novo heterozygous variant in the TRIP12 gene. Haploinsufficiency of TRIP12 has been reported to cause childhood-onset neurodevelopmental disorder including intellectual disability [29, 30], as important information cannot be obtained by ultrasound diagnosis alone. The combination of ultrasound imaging with WES-based gene variation detection could avoid the misdetection of fetal anomaly. Nevertheless, without adequate postnatal follow-up, the clinical significance of the TRIP12 gene variant was uncertain.
Remarkably, a compound heterozygous mutation of pathogenic CNV and SNV in the SMPD4 gene was identified with the performance of the CNV calling algorithm in WES data analysis (usually for SNV/Indel analysis). This suggested that the WES-based CNV calling tool could be applied as an effective way to improve the diagnostic quality and develop WES as a more powerful method in prenatal diagnosis.
This study reported 14 novel variants with a clinical significance of pathogenic or likely pathogenic among the diagnostic cases. Our study suggests that WES can broaden the mutation spectrum of genes associated with fetal structural anomalies, which would further benefit the prenatal diagnosis and genetic counselling in the future. WES also has the potential to detect new causative genes for specific developmental disorders and to establish new fetal phenotype-genotype correlations in the remaining cases without genetic diagnosis [31].
Comparing our results with eight other recent studies on fetal structural abnormalities showed that FGFR3 and COL1A1 are the most common pathogenic genes in musculoskeletal disorders, suggesting a strong genotype-phenotype correlation. Although some pathogenic genes recurred in the test results of different cohorts, the most pathogenic genes are only reported in a single study. The results indicate that the pathogenic molecular mechanism is complicated with a high degree of genetic heterogeneity and that a complete gene network needs to be discovered. WES gene variation should to be further investigated by combining of prenatal and postnatal phenotyping to provide reliable genetic diagnosis [32, 33].
Conclusions
In this study, WES was used to identify pathogenic gene mutations in fetuses with a wide range of structural anomalies. Our study will not only widen the mutation spectrum of pathogenic gene variations causing fetal structural anomalies, but also provide valuable information for prenatal counselling.
Data Availability
The details of the variant analyzed during the current study are deposited in the ClinVar repository, which is searchable by searching for an SCV accession provided in the summary report. A summary report of our successfully processed data: https://submit.ncbi.nlm.nih.gov/api/2.0/files/cpdivd1g/sub13018827__100__submitter_report_b.txt/?format=attachment. The whole-exome sequencing raw data for the participants are not publicly available due to privacy and ethical restrictions. Other researchers and readers can access these data by requesting from the corresponding author privately.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- CMA:
-
Chromosomal microarray analysis
- CNVs:
-
Copy number variants
- HPO:
-
Human phenotype ontology
- SNV:
-
Single nucleotide variant
- VUS:
-
Variants of uncertain significance
- WES:
-
Whole-exome sequencing
- XL:
-
X-linked
References
Persson M, Cnattingius S, Villamor E, Söderling J, Pasternak B, Stephansson O, et al. Risk of major congenital malformations in relation to maternal overweight and obesity severity: cohort study of 1.2 million singletons. BMJ. 2017;357:j2563.
Stanley KE, Giordano J, Thorsten V, Buchovecky C, Thomas A, Ganapathi M, et al. Causal genetic variants in Stillbirth. N Engl J Med. 2020;383:1107–16.
Ahman A, Axelsson O, Maras G, Rubertsson C, Sarkadi A. Lindgren P Ultrasonographic fetal soft markers in a low-risk population: prevalence, association with trisomies and invasive tests. Acta Obstet Gynecol Scand. 2014;93:367–73.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–11.
Aggarwal S. Role of whole exome sequencing for unidentified genetic syndromes. Curr Opin Obstet Gynecol. 2021;33:112–22.
Carss KJ, Hillman SC, Parthiban V, McMullan DJ, Maher ER, Kilby MD, et al. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet. 2014;23:3269–77.
Drury S, Williams H, Trump N, Boustred C, Lench N, Scott RH, et al. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn. 2015;35:1010–7.
Schluth-Bolard C, Diguet F, Chatron N, Rollat-Farnier PA, Bardel C, Afenjar A, et al. Whole genome paired-end sequencing elucidates functional and phenotypic consequences of balanced chromosomal rearrangement in patients with developmental disorders. J Med Genet. 2019;56:526–35.
Shi L, Li M, Qi H, Zhu J, Yang J, Tang J, et al. Whole-exome sequencing analysis to identify novel potential pathogenetic mutations in fetuses with abnormal brain structure. Ann Transl Med. 2021;9:807.
Shi X, Tang H, Lu J, Yang X, Ding H. Wu J Prenatal genetic diagnosis of omphalocele by karyotyping, chromosomal microarray analysis and exome sequencing. Ann Med. 2021;53:1285–91.
Vora NL, Powell B, Brandt A, Strande N, Hardisty E, Gilmore K, et al. Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet Med. 2017;19:1207–16.
Mellis R, Oprych K, Scotchman E, Hill M, Chitty LS. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: a systematic review and meta-analysis. Prenat Diagn. 2022;42:662–85.
Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747–57.
Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758–67.
Diderich KEM, Romijn K, Joosten M, Govaerts LCP, Polak M, Bruggenwirth HT, et al. The potential diagnostic yield of whole exome sequencing in pregnancies complicated by fetal ultrasound anomalies. Acta Obstet Gynecol Scand. 2021;100:1106–15.
Fu F, Li R, Li Y, Nie ZQ, Lei T, Wang D, et al. Whole exome sequencing as a diagnostic adjunct to clinical testing in fetuses with structural abnormalities. Ultrasound Obstet Gynecol. 2018;51:493–502.
He M, Du L, Xie H, Zhang L, Gu Y, Lei T, et al. The added value of whole-exome sequencing for anomalous fetuses with detailed prenatal ultrasound and postnatal phenotype. Front Genet. 2021;12:627204.
Normand EA, Braxton A, Nassef S, Ward PA, Vetrini F, He W, et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected mendelian disorder. Genome Med. 2018;10:74.
Vora NL, Gilmore K, Brandt A, Gustafson C, Strande N, Ramkissoon L, et al. An approach to integrating exome sequencing for fetal structural anomalies into clinical practice. Genet Med. 2020;22:954–61.
Abou Tayoun A. Mason-Suares H considerations for whole exome sequencing unique to prenatal care. Hum Genet. 2020;139:1149–59.
Best S, Wou K, Vora N, Van der Veyver IB, Wapner R, Chitty LS. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn. 2018;38:10–9.
Jelin AC, Vora NW. Exome sequencing: applications in prenatal Genetics. Obstet Gynecol Clin North Am. 2018;45:69–81.
Magini P, Smits DJ, Vandervore L, Schot R, Columbaro M, Kasteleijn E, et al. Loss of SMPD4 causes a developmental disorder characterized by Microcephaly and congenital arthrogryposis. Am J Hum Genet. 2019;105:689–705.
Simonis N, Migeotte I, Lambert N, Perazzolo C, de Silva DC, Dimitrov B, et al. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J Med Genet. 2013;50:585–92.
Chen M, Chen J, Wang C, Chen F, Xie Y, Li Y, et al. Clinical application of medical exome sequencing for prenatal diagnosis of fetal structural anomalies. Eur J Obstet Gynecol Reprod Biol. 2020;251:119–24.
Daum H, Meiner V, Elpeleg O. Harel T fetal exome sequencing: yield and limitations in a tertiary referral center. Ultrasound Obstet Gynecol. 2019;53:80–6.
Yates CL, Monaghan KG, Copenheaver D, Retterer K, Scuffins J, Kucera CR, et al. Whole-exome sequencing on deceased fetuses with ultrasound anomalies: expanding our knowledge of genetic Disease during fetal development. Genet Med. 2017;19:1171–8.
McDonald EJ, De Jesus O. StatPearls (StatPearls Publishing Copyright © 2022. StatPearls Publishing LLC.; 2022.
Bramswig NC, Lüdecke HJ, Pettersson M, Albrecht B, Bernier RA, Cremer K, et al. Identification of new TRIP12 variants and detailed clinical evaluation of individuals with non-syndromic intellectual disability with or without autism. Hum Genet. 2017;136:179–92.
Zhang J, Gambin T, Yuan B, Szafranski P, Rosenfeld JA, Balwi MA, et al. Haploinsufficiency of the E3 ubiquitin-protein ligase gene TRIP12 causes intellectual disability with or without autism spectrum disorders, speech delay, and dysmorphic features. Hum Genet. 2017;136:377–86.
Dai L, Li J, Xie L, Wang W, Lu Y, Xie M, et al. A biallelic frameshift mutation in Nephronectin causes bilateral renal agenesis in humans. J Am Soc Nephrol. 2021;32:1871–9.
Aarabi M, Sniezek O, Jiang H, Saller DN, Bellissimo D, Yatsenko SA, et al. Importance of complete phenotyping in prenatal whole exome sequencing. Hum Genet. 2018;137:175–81.
Aoi H, Mizuguchi T, Suzuki T, Makino S, Yamamoto Y, Takeda J, et al. Whole exome sequencing of fetal structural anomalies detected by ultrasonography. J Hum Genet. 2021;66:499–507.
Acknowledgements
The authors thank the patients and their families for their participation in this study.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Yayun Qin: Data analysis, Manuscript writing. Yanyi Yao, Nian Liu, Bo Wang, Lijun Liu: Data collection. Hui Li, Tangxinzi Gao, Runhong Xu, Xiaoyan Wang, Fanglian Zhang: Data analysis. Jieping Song: Data collection, Data analysis, Manuscript writing and editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance to the Declaration of Helsinki and was approved by the institutional review board of the ethics committee in the Maternal and Child Health Hospital of Hubei Province. All parents of the fetuses agreed to participate in the study and provided signed informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qin, Y., Yao, Y., Liu, N. et al. Prenatal whole-exome sequencing for fetal structural anomalies: a retrospective analysis of 145 Chinese cases. BMC Med Genomics 16, 262 (2023). https://doi.org/10.1186/s12920-023-01697-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01697-3