
Abstract
Background
PTEN hamartoma syndrome (PHTS) is an autosomal dominant disorder characterized by pathogenic variants in the tumor suppressor gene phosphatase and tensin homolog (PTEN). It is associated with an increased risk of muco-cutaneous features, hamartomatous tumors, and cancers. Mosaicism has been found in a few cases of patients with de novo PHTS, identified from blood samples. We report a PHTS patient with no variant identified from blood sample. Constitutional PTEN mosaicism was detected through sequencing of DNA from different tumoral and non-tumoral samples.
Case presentation
Our patient presented clinical Cowden syndrome at 56 years of age, with three major criteria (macrocephaly, Lhermitte Duclos disease, oral papillomatosis), and two minor criteria (structural thyroid lesions, esophageal glycogenic acanthosis). Deep sequencing of PTEN of blood leukocytes did not reveal any pathogenic variants. Exploration of tumoral (colonic ganglioneuroma, esophageal papilloma, diapneusia fibroids) and non-tumoral stomach tissues found the same PTEN pathogenic variant (NM_000314.4 c.389G > A; p.(Arg130Gln)), with an allelic frequency of 12 to 59%, confirming genomic mosaicism for Cowden syndrome.
Conclusions
This case report, and review of the literature, suggests that systematic tumor analysis is essential for patients presenting PTEN hamartoma syndrome in the absence of any causal variant identified in blood leukocytes, despite deep sequencing. In 65 to 70% of cases of clinical Cowden syndrome, no pathogenic variant in the PTEN is observed in blood samples: mosaicism may explain a significant number of these patients. Tumor analysis would improve our knowledge of the frequency of de novo variations in this syndrome. Finally, patients with mosaicism for PTEN may not have a mild phenotype; medical care identical to that of heterozygous carriers should be offered.
Similar content being viewed by others

Background
PTEN hamartoma syndrome (PHTS) is an autosomal dominant disorder characterized by pathogenic variants in the tumor suppressor gene phosphatase and tensin homolog (PTEN) [1]. The clinical presentation is heterogeneous, including Cowden syndrome (CS) [2], Bannayan-Riley-Ruvalcaba syndrome (BRRS) [3], Lhermitte–Duclos disease [4], Segmental outgrowth-lipomatosis-arteriovenous malformation-epidermal nevus (SOLAMEN) syndrome [5], and autism-macrocephaly syndrome (ASD) [6].
The prevalence of CS, probably underestimated, is estimated at 1 in 200,000 individuals [7], with a penetrance of up to 90% in the second decade [8]. It is associated with an increased risk of muco-cutaneous features, hamartomatous tumors, and cancers (Table 1) [9,10,11].
Mosaicism has been found in a few cases of patients with de novo PHTS [13]. This mosaicism can sometimes be identified in blood samples, using different techniques including deep next generation sequencing (NGS).
We present a patient with clinical CS, with no identified pathogenic variation in PTEN through deep sequencing of DNA from blood samples. Constitutional PTEN mosaicism was detected through sequencing of DNA from different tumoral and non-tumoral tissues. We review PHTS mosaicism reported in the literature, and suggest that mosaicism be actively searched for when possible.
Case presentation
A 56-year-old man presented left-sided hyperacusis and loss of ipsilateral stapedial reflex. A cerebral MRI was performed and identified an infiltrative mass 57 mm in diameter located in the left cerebellar hemisphere, without enhancement of the contrast product, suggestive of a hamartomatous lesion of the posterior fossa.
Given the absence of symptoms, in particular of intracranial hypertension on ophthalmological examination, a control MRI was performed 3 months later. This second MRI found a characteristic appearance of Lhermitte Duclos disease (dysplastic gangliocytoma) of the left cerebellar hemisphere, with a striated lamellar appearance, measuring 60 mm × 36 mm × 31 mm. This lesion was associated with a mass effect on the lower part of the fourth ventricle, resulting in moderate triventricular hydrocephalus (Evans Index at 0.35), with slight signs of transpedicular cerebrospinal fluid (CSF) resorption.
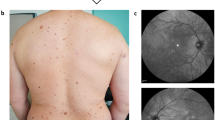
The patient’s medical records did not report any delay in psychomotor development or learning difficulties. He had a history of “diapneusia fibroids” of the tongue that had been treated surgically some years earlier, and high blood pressure which was well-controlled by a combination of Angiotensin II antagonist and calcium channel blocker. The clinical examination found macrocephaly, with a head circumference of 60 cm (+ 2SD). Cutaneous-mucosal examination identified seborrheic keratosis in the shoulders and in the extremities. It also found fibrous lesions in interdental positions 31–32 and 32–33, as well as on the right lateral edge of the tongue. No cutaneous trichilemmomas or papules were found.
In this context, a check-up for other lesions was performed. Thyroid ultrasound found multinodular hypertrophy of the right lobe with five nodular and cystic lesions (3 to 15 mm in diameter), Eu-thirads 3. Esogastroduodenal fibroscopy found oesophageal papillomatosis, confirmed histologically, and hyperplastic polyps of the gastric antrum. Colonoscopy identified a 6-mm-diameter ganglioneuroma of the transverse colon, confirmed histologically.
The patient’s family history was not suggestive of PHTS, with no notable history. A clinical diagnosis of CS was made, due to the presence of three major criteria: Lhermitte Duclos disease in adult, macrocephaly, and oral papillomatosis. Two minor criteria were also present, including thyroid structural lesions and esophageal glycogenic acanthosis, as well as a unique digestive ganglioneruoma.
Methods
To confirm the clinical diagnosis, constitutional and tumor analyses of the PTEN gene (NM_000314.4) were performed.
Samples
DNA was extracted from peripheral blood using the QIAamp DNA Blood maxikit (Qiagen) and from oesophageal papilloma, ganglioneuroma, diapneusia fibroid and non-tumoral stomach tissue formalin-fixed paraffin-embedded blocks, using the Maxwell 16 FFPE Plus LEV kit (Promega). Libraries were prepared using KAPA HyperPlus Kits and captured using KAPA HyperCap Target Enrichment (Roche). The quality of libraries and captures was controlled with a Tapestation 4150 (Agilent). Sequencing was performed with a NextSeq 500/550 High Output Kit v2.5 (300 Cycles) on a NextSeq 550 instrument (Illumina). The percentage of tumor cells from the colon sample was 60%. The tumor surface of the esophageal sample, diapneusia fibroid and non-tumoral stomach tissue was respectively 16 mm2, 6 mm2 and 2 mm2.
Bio-informatic analysis
De-multiplexing was performed using bcl2fastq2 Conversion Software (Illumina). Alignment was performed on University of California Santa Cruz human genome reference build 19 using the Burrows-Wheeler Aligner (v0.7.17). Genome Analysis Toolkit (GATK v 4.1.8.0) and PICARD (v 2.22.8) tools were used for base quality score recalibration (BaseRecalibrator) and identified duplicate reads (MarkDuplicates). Variant calling was performed by GATK HaplotypeCaller and by GATK Mutect and Freebayes (v1.2.0). Annotations were added by Ensembl VariantEffectPredictor (v96.0) and Annovar (8 Jun 2020). Copy number variation analysis was performed using ExomeDepth (v 1.1.12).
The entire coding sequence and intron/exon junctions (positions up to -20 and + 6) of PTEN gene were analyzed in all samples (blood, tumoral and non-tumoral). The entire 5'UTR and 3'UTR regions up to position *20 were analyzed from blood samples only. Due to the clinical presentation, no other gene was analyzed from all samples explored. Variants were filtered on mapping quality, call quality and minimum depth of coverage of 30 × for blood sample and 200 × for tumor samples. The detection threshold for constitutional analysis was 20% of reads. The detection threshold for tumor analyses was 10% of reads. The technical sensitivity was greater than 95%.
Variant interpretation was performed using ALAMUT Visual 2.11 (Interactive BioSoftware), and relevant databases (ClinVar). Splice variants were interpreted using SpliceAI (v1.3) and SPIP (v1.0) softwares. Variants were classified according to the American College of Medical Genetics (ACMG) recommendations.
Results
NGS sequencing from two independent blood samples was first performed. The average depth for PTEN was 1633 with 100% coverage at 30X, 98.5% at 100X and 77.61% at 1000X. No constitutional variation of the PTEN gene was found from blood samples.
Given the strong clinical suspicion, and the absence of a probable pathogenic or pathogenic variant identified in PTEN from the blood samples, two tumor samples were analyzed (oesophageal papilloma, ganglioneuroma). The average depth was 3807 with 100% coverage. at 1000X and 95% at 1500X. The same pathogenic variant, c.389G > A; p.(Arg130Gln), was identified in both samples. The variant allele frequency was 12% in the ganglioneuroma sample (tumor cell content 60%) and 59% in the esophageal papilloma sample (tumor cell content unknown) (Fig. 1). To confirm the presence of constitutional mosaic PTEN in the patient and rule out the hypothesis that the same mutation occurred in two different tissues in an acquired process, two supplemental samples were expored, including one non-tumoral sample (diapneusia fibroid and non-tumoral stomach tissue). The same pathogenic variant was found with an allele frequency respectively at 25% and 14%, confirming the diagnosis.

Variant c.389G > A; p.(Arg130Gln) found in mosaic in two tumoral sample in PTEN gene (NM_000314.4)
This variant is located in the phosphatase functional domain of the protein. It is a recurrent pathogenic variation, involved in different clinical presentations associated with PTEN hamartoma tumor syndrome.
In addition, an acquired unknown significant variant c.816C > G, p.(His272Gln) was found in ganglioneuroma and papilloma tumoral samples, with allelic frequency respectively at 11 and 8%.
Discussion and conclusions
Mosaicism is very rare in PTHS and only a few cases have been published. Most of these were reported in the study of Rofes et al.[13], that included six patients, and three another cases were reported in the studies of Steffan et al. and Hendricks et al. [13,14,15,16,17,18,19,20] (Table 2).
As with heterozygous constitutional PTEN variants, the entire clinical spectra of PTEN can be present as a mosaic, including Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, autism-macrocephaly syndrome and Lhermitte–Duclos disease [21]. PTEN mosaicism does not seem to be associated with attenuated forms of the pathology: all reported patients except one, and including ours, presented validated clinical criteria. Thus, it is very difficult in practice to distinguish PTEN mosaicism from a priori de novo PHTS.
The prevalence of detected constitutional PTEN variations in patients meeting diagnostic criteria of CS or BRRS is only 30–35% [12]. Moreover, the frequency of de novo variations is significant in this syndromes, estimated at 10.7 to 47.6% of cases [22]. Thus, the absence of identification of variation of interest in patients with CS, in a context of frequent de novo mutations, could be explained using different hypotheses. Firstly, genes other than PTEN could be involved. Alternatively, mosaicism for a causal mutation in the PTEN gene may not be detectable using routine techniques and approaches.
Concerning the first hypothesis, various studies have identified candidate genes of interest, such as KLLN, AKT1, PIK3CA [23], SEC23B [24] or more recently WWP1 [25]. Although the involvement of these genes was not confirmed at this time by independent laboratories, their exploration may be of interest, with the exception of AKT1 which remains associated with a particular phenotype (Proteus syndrome), in patients with PTHS criteria not carrying a causal pathogenic variant in PTEN, which is not the case in our study.
The second hypothesis depends on the samples analyzed and the analysis techniques. Most of the patients reported with constitutional PTEN mosaicism were diagnosed based on the analysis of blood samples. The classical Sanger technique is sensitive enough to find mosaicism with an allelic frequency of about 10%, which was enough to confirm the diagnosis in a number of cases [26].
However, for low-level mosaicisms, NGS and deep sequencing exploration is preferred and it is now performed routinely in many molecular genetics laboratories. The improved sensitivity allowed detection of variants at lower allele frequencies in two PHTS patients of the cohort (1.70% and 3.50% from lymphocytes). In our patient, NGS sequencing failed to detect a variant in DNA from blood, and tumor samples exploration was required. After the identification of the same variant in different tumors and healthy tissue, manual proofreading of the blood sequencing data revealed the causal PTEN variant at a frequency of 0.7% (17 / 2285 reads) which is below reasonable sensitivity in diagnostic practice. A similar patient, with a variant undetectable in the blood sample (VAF < 1%) but found in two other tissues (buccal swab, venous malformation), was reported by Hendricks et al. [20].
Deep sequencing (≥ 2000x) in the exploration of mosaicism in hereditary predisposition to cancer is of well-known interest and can detect single nucleotide variants at a frequency of 2–3% in blood samples [26]. However, our study underlines that its use in clinical practice remains difficult in cases with very low mosaicism in blood samples. Indeed, variants with lower allele frequency could only be explored with manual inspection of the aligned reads at hotspots or known positions identified in other samples, as in our patient. Moreover, the detection of variants with very low allele frequency could originate from circulating tumor DNA or a sequencing error. Their interpretation in a diagnostic approach to confirm or not the presence of mosaicism does not therefore seem to be reasonable in routine practice.
Thus, the absence of a pathogenic variant in PTEN from blood sample does not rule out the diagnosis of PHTS, and the continued investigation of other tissues must be systematic in patients presenting the testing criteria. This is relevant in this syndrome, because multiple tumoral occurrences are very common. A non-tumor sample could also help to confirm the molecular diagnosis. This is what was done in our study, with the exploration of a non-tumoral stomach tissue sample, confirming the presence of the pathogenic variant PTEN outside any context of tumorogenesis, directing on its constitutional character. It should be noted that the cellular composition of buccal swab samples tends to be rich in leukocytes, and should not be considered a different that blood sample [27].
Mosaicism could explain some patients presenting diagnostic criteria for PTEN analysis, without identified causal variation. Systematic tumor analysis for these cases, as well as a cohort study, could better estimate the frequency of mosaicism in PHTS. The identification of these patients is important in clinical practice, for their medical management, as well as for that of their family.
Because PTEN mosaicism does not seem to be associated with a milder phenotype, the clinical management of concerned patients, including genetic counselling and pre-natal testing, should be the same as that for people with a constitutional heterozygous variant, with the exception of parental testing, which is not useful in the context.
In conclusion, we report a case of PTEN mosaicism in a patient diagnosed through DNA sequencing from tumoral and non-tumoral tissue samples. This report highlights the interest of additional tissue analysis when peripheral blood sample DNA sequencing does not identify a causal variant. Based on a review of the literature, we propose that this analysis should now be systematic in this context in clinical practice. Moreover, the literature review provided in this report suggests that PTEN mosaicism is not associated with a milder phenotype of the syndrome. The personal and family care of these patients should be identical to that proposed to heterozygous carriers of PTEN, with the exception of testing of the parents.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACMG:
-
American College of Medical Genetics
- CSF:
-
Cerebrospinal fluid
- GATK:
-
Genome Analysis Toolkit
- NGS:
-
Next generation sequencing
- PHTS:
-
PTEN hamartoma syndrome
- PTEN :
-
Phosphatase and tensin homolog
- SOLAMEN:
-
Segmental outgrowth-lipomatosis-arteriovenous malformation-epidermal nevus
References
Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16(11):1289–300.
Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns févr. 2009;18(1):13–27.
Lachlan KL, Lucassen AM, Bunyan D, Temple IK. Cowden syndrome and Bannayan–Riley–Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J Med Genet sept. 2007;44(9):579–85.
Zhou XP, Marsh DJ, Morrison CD, Chaudhury AR, Maxwell M, Reifenberger G, et al. Germline Inactivation of PTEN and dysregulation of the Phosphoinositol-3-Kinase/Akt pathway cause human Lhermitte-Duclos disease in adults. Am J Hum Genet. 2003;73(5):1191–8.
Caux F, Plauchu H, Chibon F, Faivre L, Fain O, Vabres P, et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet juill. 2007;15(7):767–73.
Butler M, Dasouki M, Zhou X, Talebizadeh Z, Brown M, Takahashi T, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet avr. 2005;42(4):318–21.
Hendricks LAJ, Hoogerbrugge N, Mensenkamp AR, Brunet J, Lleuger-Pujol R, Høberg-Vetti H, et al. Cancer risks by sex and variant type in PTEN hamartoma tumor syndrome. J Natl Cancer Inst. 2023;115(1):93–103.
Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol. Methods mai. 2015;77–78:11–9.
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18(2):400–7. https://doi.org/10.1158/1078-0432.CCR-11-2283.
Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105(21):1607–16.
Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet avr. 2013;50(4):255–63.
Pilarski R. PTEN Hamartoma tumor syndrome: a clinical overview. Cancers (Basel). 2019;11(6):844.
Rofes P, Teulé Á, Feliubadaló L, Salinas M, Cuesta R, Iglesias S, et al. Mosaicism in PTEN—new case and comment on the literature. Eur J Hum Genet. 2022;30:1–4.
Steffann J, Michot C, Borghese R, Baptista-Fernandes M, Monnot S, Bonnefont JP, et al. Parental mosaicism is a pitfall in preimplantation genetic diagnosis of dominant disorders. Eur J Hum Genet. 2014;22(5):711–2.
Gammon A, Jasperson K, Pilarski R, Prior T, Kuwada S. PTEN mosaicism with features of Cowden syndrome. Clin Genet. 2013;84(6):593–5.
Pritchard CC, Smith C, Marushchak T, Koehler K, Holmes H, Raskind W, et al. A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Genet Med déc. 2013;15(12):1004–7.
Salo-Mullen EE, Shia J, Brownell I, Allen P, Girotra M, Robson ME, et al. Mosaic partial deletion of the PTEN gene in a patient with Cowden syndrome. Familial Cancer. 2014;13(3):459–67.
Golas MM, Auber B, Ripperger T, Pabst B, Schmidt G, Morlot M, et al. Looking for the hidden mutation: Bannayan–Riley–Ruvalcaba syndrome caused by constitutional and mosaic 10q23 microdeletions involving PTEN and BMPR1A. Am J Med Genet A. 2019;179(7):1383–9.
Goldenberg A, Marguet F, Gilard V, Cardine AM, Hassani A, Doz F, et al. Mosaic PTEN alteration in the neural crest during embryogenesis results in multiple nervous system hamartomas. Acta Neuropathol Commun. 2019;7:191.
Hendricks LAJ, Schuurs-Hoeijmakers J, Spier I, Haadsma ML, Eijkelenboom A, Cremer K, et al. Catch them if you are aware: PTEN postzygotic mosaicism in clinically suspicious patients with PTEN Hamartoma Tumour syndrome and literature review. Eur J Med Genet juill. 2022;65(7): 104533.
Cavaillé M, Ponelle-Chachuat F, Uhrhammer N, Viala S, Gay-Bellile M, Privat M, et al. Early onset multiple primary tumors in atypical presentation of Cowden syndrome identified by whole-exome-sequencing. Front Genet. 2018;9:353.
Mester J, Eng C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet Med sept. 2012;14(9):819–22.
Yehia L, Eng C. PTEN hamartoma tumour syndrome: what happens when there is no PTEN germline mutation? Hum Mol Genet. 2020;29:R150–7.
Yehia L, Niazi F, Ni Y, Ngeow J, Sankunny M, Liu Z, et al. Germline Heterozygous variants in SEC23B are associated with Cowden syndrome and enriched in apparently sporadic thyroid cancer. Am J Hum Genet. 2015;97(5):661–76.
Lee YR, Yehia L, Kishikawa T, Ni Y, Leach B, Zhang J, et al. WWP1 Gain-of-function inactivation of PTEN in cancer predisposition. N Engl J Med. 2020;382(22):2103–16.
Steinke-Lange V, de Putter R, Holinski-Feder E, Claes KBM. Somatic mosaics in hereditary tumor predisposition syndromes. Eur J Med Genet. 2021;64: 104360.
Theda C, Hwang SH, Czajko A, Loke YJ, Leong P, Craig JM. Quantitation of the cellular content of saliva and buccal swab samples. Sci Rep. 2018;8:6944. https://doi.org/10.1038/s41598-018-25311-0.
Acknowledgements
Thanks to the Centre Jean Perrin for supporting this study.The samples used in this study were conserved in the Biological Resource Center of Jean Perrin Comprehensive Cancer Center, identified under No. BB-0033-00075 (Clermont-Ferrand, France).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors have read and approved the manuscript. MC, and YJB designed the study. MC, FP-C, VB, NJ, performed the acquisition of data. MC, NU, ZO, YB, VB, NJ, NS, YJB, DC, VA performed analysis, interpretation of data, and critically revised the article. MC drafted the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study has been approved by local ethics committee (CPP Sud-Est VI Clermont-Ferrand); reference: IRB: 2022 / CE 42.
Consent for publication
The patient give his written consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cavaillé, M., Crampon, D., Achim, V. et al. Diagnosis of PTEN mosaicism: the relevance of additional tumor DNA sequencing. A case report and review of the literature. BMC Med Genomics 16, 166 (2023). https://doi.org/10.1186/s12920-023-01600-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01600-0