
Abstract
Background
Xq22.1–q22.3 deletion is a rare chromosome aberration. The purpose of this study was to identify the correlation between the phenotype and genotype of chromosome Xq22.1–q22.3 deletions.
Methods
Chromosome aberrations were identified by copy number variation sequencing (CNV-seq) technology and karyotype analysis. Furthermore, we reviewed patients with Xq22.1–q22.3 deletions or a deletion partially overlapping this region to highlight the rare condition and analyse the genotype–phenotype correlations.
Results
We described a female foetus who is the “proband” of a Chinese pedigree and carries a heterozygous 5.29 Mb deletion (GRCh37: chrX: 100,460,000–105,740,000) in chromosome Xq22.1–q22.3, which may affect 98 genes from DRP2 to NAP1L4P2. This deletion encompasses 7 known morbid genes: TIMM8A, BTK, GLA, HNRNPH2, GPRASP2, PLP1, and SERPINA7. In addition, the parents have a normal phenotype and are of normal intelligence. The paternal genotype is normal. The mother carries the same deletion in the X chromosome. These results indicate that the foetus inherited this CNV from her mother. Moreover, two more healthy female family members were identified to carry the same CNV deletion through pedigree analysis according to the next-generation sequencing (NGS) results. To our knowledge, this family is the first pedigree to have the largest reported deletion of Xq22.1–q22.3 but to have a normal phenotype with normal intelligence.
Conclusions
Our findings further improve the understanding of the genotype–phenotype correlations of chromosome Xq22.1–q22.3 deletions.This report may provide novel information for prenatal diagnosis and genetic counselling for patients who carry similar chromosome abnormalities.
Similar content being viewed by others

Background
Birth defects are a major public health problem that lead to miscarriage, foetal death, premature birth and childhood disabilities [1]. In China, approximately 5.6% of newborns are affected by birth defects annually; of these, chromosome aberrations account for more than 80% of the genetic causes, including abnormalities in chromosome number (aneuploidy) or structure, large fragment deletion/duplication, and pathogenic copy number variations (CNVs) [2, 3]. With the implementation of the universal “two child” policy, the proportion of birth defects has increased. This increase might be partly due to the increase in maternal age at delivery, the proportion of mothers with complications, and the number of multiple pregnancies. However, the increase in the number of prenatal screening or prenatal diagnoses for pregnant women of advanced age in China might have alleviated this increasing trend in birth defects [4].
For decades, karyotype analysis has been widely used as the “gold standard” for chromosome aberrations, as it can identify aneuploidy, translocation and inversion of chromosomes. However, karyotyping cannot detect abnormalities in chromosome fragments smaller than 5–10 Mb. Notably, more than 300 types of microdeletion/microduplication syndromes that are caused by CNVs smaller than 5 Mb have been identified, and they account for half of the birth defects caused by chromosome aberrations. CNV sequencing (CNV-seq) technology has brought opportunities and challenges to the detection of chromosome aberrations smaller than 5 Mb. In 2019, genetic experts suggested that CNV-seq could be used as a first-line prenatal diagnosis test for pregnant women who may have foetal chromosome abnormalities in China [5, 6].
Large fragment deletions in chromosome Xq22 might cause neurodevelopmental disorders, including severe intellectual disability and behavioural abnormalities. In this study, we report a female foetus who carries a heterozygous 5.29 Mb deletion in chromosome Xq22.1–q22.3 (including 7 known morbid genes), which was inherited from her healthy mother who had a normal phenotype with normal intelligence.
Methods
Karyotype analysis
The pregnant women underwent amniocentesis for karyotype analysis to identify chromosome aberrations of the foetus. In addition, karyotype analysis of peripheral blood were performed in the nonconsanguineous parents to determine the possible causes of chromosome aberration. Using conventional G-banding analysis technology, twenty-five metaphases were analysed at the 550 chromosome band resolution.
CNV sequencing analysis
CNV sequencing procedures, including DNA extraction, library construction, next-generation sequencing (NGS), bioinformatics analysis, and quality control (QC), were performed in our NGS laboratory with the Ion Torrent platform (BioelectronSeq 4000 sequencing system: Life Technologies, USA) according to the manufacturer’s protocol (Product No. S30030).
Results
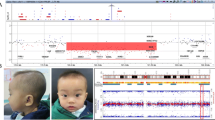
A healthy pregnant woman, who was 37 years old with G3P1A1, had a 12-year-old healthy daughter. The pregnant woman requested prenatal diagnosis due to advanced maternal age. 3D ultrasound examination showed no evidence of foetal anomalies. She underwent amniocentesis for karyotype analysis and CNV-seq at 23 + 5 weeks gestation at Taizhou Hospital of Zhejiang Province. The foetal karyotype analysis showed a normal female karyotype of 46,XX (Additional file 1). However, the results of CNV-seq analysis indicated a 5.29 Mb deletion in chromosome Xq22.1–q22.3 (GRCh37/hg19: chrX: 100,460,000–105,740,000), which may affect 98 genes from DRP2 to NAP1L4P2 according to the Ensembl genome browser (https://asia.ensembl.org/) (Fig. 1) and the ClinGen database (https://www.clinicalgenome.org/) (Additional file 2). According to the DECIPHER database (https://www.deciphergenomics.org), this CNV deletion encompassed 42 OMIM genes, including DRP2, TAF7L, TIMM8A, BTK, RPL36A, GLA, HNRNPH2, ARMCX1 ~ 6, ZMAT1, BEX1 ~ 5, NXF2 ~ 5, TMSB15A, GPRASP1 ~ 2, BHLHB9, RAB40AL, TCEAL7, RAB40A, TCEAL1, MORF4L2, PLP1, RAB9B, TMSB15B, H2BW1, SLC25A53, ESX1, IL1RAPL2, TEX13A, NRK, and SERPINA7 [7]. Among these OMIM genes, there are 7 known morbid genes, including TIMM8A, BTK, GLA, HNRNPH2, GPRASP2, PLP1, and SERPINA7. In addition, the nonconsanguineous parents have a normal phenotype and are of normal intelligence. Their intellectual levels have not been precisely tested, but they judged to be normal from their normal social activities.

Ensembl genome browser image showing the Xq22.1–q22.3 deletions (GRCh37: ChrX: 100,460,000–105,740,000). Red frame indicate the location of the deletion regions identified in the Chinese pedigree in this study
When the pregnant woman had genetic counselling in our prenatal diagnosis centre, we learned that she had a term birth of a healthy girl in 2010 and suffered a termination of pregnancy due to the 46, XXX karyotype of the foetus in 2018. The family wanted to know whether the foetus would have genetic defects after birth. Therefore, we further investigated this pedigree to determine the possible causes of the Xq22.1–q22.3 deletion (Fig. 2). Is it due to parental inheritance or a novel foetal mutation? Further pedigree analysis indicated that the CNV deletion of this foetus was inherited from her healthy mother. Moreover, two more healthy female family members (the pregnant woman’s daughter and mother) were identified to carry the same Xq22.1–q22.3 deletion (Fig. 3). The pregnant woman has a normal clinical phenotype with regular menses and normal fertility. There were no problems during pregnancy or delivery. Her daughter is now 12 years old with normal physical and psychomotor development. Her mother is now 65 years old with normal physical and psychomotor development. Through a genotype–phenotype correlation analysis, although the 5.29 Mb deletion in chromosome Xq22.1–q22.3 was inherited from a normal phenotype parent, it is still considered to be a pathogenic CNV in this pedigree as it contains 7 known morbid genes (TIMM8A, BTK, GLA, HNRNPH2, GPRASP2, PLP1, and SERPINA7).

Three-generation pedigree of a Chinese family and carries a heterozygous 5.29 Mb deletion in chromosome Xq22.1–q22.3

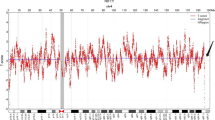
Chromsomal aberrations revealed by CNV-seq analysis are shown with Agilent Genomic Workbench (Agilent Technologies) in chromosome view. X- and Y-axes indicate chromosomal location and signal log2 ratio, respectively. A microdeletion is shown in Xq22.1–q22.3 region (5.29 Mb)
After genetic counselling, the couple decided to continue with the pregnancy. On February 28, 2022, a female neonate weighing 4.4 kg and 49 cm in length was born at 39 plus 3 weeks of pregnancy by spontaneous labour. The foetus had a five-minute Apgar score of 10 points, and no abnormal clinical symptoms or signs have been observed to date.
Discussion
In this rare Chinese pedigree, no abnormality was found in the G-banding karyotype analysis of the foetus or her parents. As the “gold standard” for chromosome aberrations, conventional Giemsa-banding karyotype analysis cannot detect chromosome abnormalities at a resolution of smaller than 5–10 Mb. However, CNV-seq technology provides opportunities and challenges to detect chromosome aberrations smaller than 5 Mb. In this study, CNV-seq analysis of uncultured amniotic fluid cells showed a 5.29 Mb deletion (GRCh37: chrX: 100,460,000–105,740,000) in chromosome Xq22.1–q22.3. It appears that the 5.29 Mb deletion in Xq22.1–q22.3 is a rare chromosome aberration. The foetus inherited this CNV deletion from her healthy mother.
A total of 98 genes were mapped to this 5.29 Mb deletion CNV. This fragment encompasses 7 known morbid genes, translocase of inner mitochondrial membrane 8A (TIMM8A), Bruton tyrosine kinase (BTK), galactosidase alpha (GLA), heterogeneous nuclear ribonucleoprotein H2 (HNRNPH2), G protein-coupled receptor associated sorting protein 2 (GPRASP2), proteolipid protein 1 (PLP1), and serpin family A member 7 (SERPINA7). According to the OMIM database (http://omim.org/), defects in the TIMM8A gene are the cause of Mohr–Tranebjaerg syndrome (MTS) [MIM #304700], defects in BTK are the cause of X-linked agammaglobulinemia (XLA) [MIM #300755], defects in GLA are the cause of Fabry disease (FD) [MIM #301500], defects in HNRNPH2 are the cause of the bain type of X-linked syndromic intellectual developmental disorder (MRXSB)[MIM #300986], defects in GPRASP2 are the cause of X-linked deafness-7 (DFNX7) [MIM #301018], defects in PLP1 are the cause of Pelizaeus–Merzbacher disease (PMD) [MIM #312080] or Spastic paraplegia 2 (SPG2) [MIM #312920], and defects in SERPINA7 are the cause of Thyroxine-binding globulin quantitative trait locus (TBGQTL) [MIM #300932].
A literature review identified that more than 43 families and 56 cases involving the affected region of Xq22.1–q22.3 deletion or a deletion that partially overlaps have been previously reported [8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. None of these previously reported cases had the same CNV deletion as the Chinese pedigree we reported. As shown in Table 1, we analysed the genotype–phenotype correlations of these patients with CNV deletions in chromosome Xq22.1–q22.3. Among them, the phenotype of female cases mainly include severe mental or physical limitations [9, 12,13,14,15,16,17, 27]. But so far, only one 4-year-old female of Xq22.1 → qter deletion had a normal phenotype [32]. Fortunately, in this Chinese pedigree, all three females with the same Xq22.1–q22.3 deletion have a normal phenotype, most likely due to complete inactivation of the abnormal X chromosomes in females [14, 17]. Notably, no abnormal clinical symptoms or signs have been observed in the fourth female neonate in this Chinese pedigree to date. However, further follow-up will still be necessary to evaluate the phenotype.
In addition, we focus on the genetic patterns of these morbid genes. Mohr–Tranebjaerg syndrome is caused by mutations in the TIMM8A gene, which is a rare X-linked recessive disorder resulting in early-onset hearing impairment, progressive visual deterioration, and gradual dystonia. Some female carriers showed signs of minor neuropathy and mild hearing impairment [33, 34]. Fabry disease is a rare X-linked lipid storage disorder caused by a deficiency or absence of lysosomal alphagalactosidase A, which encoded by GLA gene. It is worth noting that heterozygous women should not be called carriers because they often been reported with a wide range of clinical symptoms. The early clinical manifestations mainly include acroparesthesias, angiokeratomas, pain crisis, and cornea verticillata, among other abnormalities. It therefore appears that Fabry disease affects both hemizygotes and heterozyotes, and should be considered an X-linked dominant disorder [35, 36]. Pelizaeus–Merzbacher disease is an X-linked recessive central nervous system disorder, which belongs to the group of hypomyelinating leukodystrophy (HLD1). PMD principally affect males and occasionally observed in carrier females, which is characterized clinically by nystagmus, spastic quadriplegia, ataxia, and developmental delay [9, 17, 37]. In addition, there was no report of male patients with large fragment Xq22 deletions. This is probably because larger Xq22 deletions may lead to embryonic lethality in males, since male patients with smaller nullisomy in the vicinity show more severe developmental delay [8, 10,11,12, 17].
Conclusions
X chromosomal deletions are infrequent findings in prenatal diagnosis and present a difficult counselling challenge when they occur. Genotype–phenotype correlation analysis can provide reliable clinical genetic counselling for chromosome abnormality reports. In addition, the X-inactivation pattern could provide an opportunity for more informative genetic counselling when a de novo CNV deletion in the X chromosome is detected.
Availability of data and materials
All data generated during this study are included in this published article.
Abbreviations
- BTK :
-
Bruton tyrosine kinase
- CNV:
-
Copy number variation
- DFNX7:
-
X-linked deafness-7
- FD:
-
Fabry disease
- GLA :
-
Galactosidase alpha
- GPRASP2 :
-
G protein-coupled receptor associated sorting protein 2
- HNRNPH2 :
-
Heterogeneous nuclear ribonucleoprotein H2
- MRXSB:
-
X-linked syndromic intellectual developmental disorder
- MTS:
-
Mohr–Tranebjaerg syndrome
- NGS:
-
Next-generation sequencing
- OMIM:
-
Online Mendelian Inheritance in Man
- PLP1 :
-
Proteolipid protein 1
- PMD:
-
Pelizaeus–Merzbacher disease
- SERPINA7 :
-
Serpin family A member 7
- SPG2:
-
Spastic paraplegia 2
- TBGQTL:
-
Thyroxine-binding globulin quantitative trait locus
- TIMM8A :
-
Translocase of inner mitochondrial membrane 8A
- XLA:
-
X-linked agammaglobulinemia
References
Harris BS, Bishop KC, Kemeny HR, Walker JS, Rhee E, Kuller JA. Risk factors for birth defects. Obstet Gynecol Surv. 2017;72(2):123–35.
Li Z, Di J. Prevention and control of birth defects in China: achievements and challenges. China CDC Wkly. 2021;3(37):771–2.
Zhou Y, Mao X, Zhou H, Wang L, Qin Z, Cai Z, Yu B. Birth defects data from population-based birth defects surveillance system in a district of Southern Jiangsu, China, 2014–2018. Front Public Health. 2020;8:378.
Zeng Y, Hesketh T. The effects of China’s universal two-child policy. Lancet. 2016;388(10054):1930–8.
Clinical genetics group, medical genetics branch, Chinese Medical Association. Expert consensus on the application of low-depth whole genome sequencing in prenatal diagnosis. Chin J Med Genet. 2019;36(4):293–6.
Wang J, Chen L, Zhou C, Wang L, Xie H, Xiao Y, Zhu H, Hu T, Zhang Z, Zhu Q, Liu Z, Liu S, Wang H, Xu M, Ren Z, Yu F, Cram DS, Liu H. Prospective chromosome analysis of 3429 amniocentesis samples in China using copy number variation sequencing. Am J Obstet Gynecol. 2018;219(3):287.e1-18.
Firth HV, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84:524–33.
Raskind WH, Williams CA, Hudson LD, Bird TD. Complete deletion of the proteolipid protein gene (PLP) in a family with X-linked Pelizaeus–Merzbacher disease. Am J Hum Genet. 1991;49(6):1355–60.
Inoue K, Osaka H, Thurston VC, Clarke JT, Yoneyama A, Rosenbarker L, Bird TD, Hodes ME, Shaffer LG, Lupski JR. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet. 2002;71(4):838–53.
Hübner CA, Orth U, Senning A, Steglich C, Kohlschütter A, Korinthenberg R, Gal A. Seventeen novel PLP1 mutations in patients with Pelizaeus–Merzbacher disease. Hum Mutat. 2005;25(3):321–2.
Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131(7):1235–47.
Torisu H, Iwaki A, Takeshita K, Hiwatashi A, Sanefuji M, Fukumaki Y, Hara T. Clinical and genetic characterization of a 2-year-old boy with complete PLP1 deletion. Brain Dev. 2012;34(10):852–6.
Matsufuji M, Osaka H, Gotoh L, Shimbo H, Takashima S, Inoue K. Partial PLP1 deletion causing X-linked dominant spastic paraplegia type 2. Pediatr Neurol. 2013;49(6):477–81.
Yamamoto T, Wilsdon A, Joss S, Isidor B, Erlandsson A, Suri M, Sangu N, Shimada S, Shimojima K, Le Caignec C, Samuelsson L, Stefanova M. An emerging phenotype of Xq22 microdeletions in females with severe intellectual disability, hypotonia and behavioral abnormalities. J Hum Genet. 2014;59(6):300–6.
Brender T, Wallerstein D, Sum J, Wallerstein R. Unusual presentation of Pelizaeus–Merzbacher disease: female patient with deletion of the proteolipid protein 1 gene. Case Rep Genet. 2015;2015: 453105.
Kinoshita M, Roston W. Rare case of female with Pelizaeus Mertzbacher disease due to deletion of proteolipid protein 1: a case report. JNMA J Nepal Med Assoc. 2018;56(214):967–9.
Hijazi H, Coelho FS, Gonzaga-Jauregui C, Bernardini L, Mar SS, Manning MA, Hanson-Kahn A, Naidu S, Srivastava S, Lee JA, Jones JR, Friez MJ, Alberico T, Torres B, Fang P, Cheung SW, Song X, Davis-Williams A, Jornlin C, Wight PA, Patyal P, Taube J, Poretti A, Inoue K, Zhang F, Pehlivan D, Carvalho CMB, Hobson GM, Lupski JR. Xq22 deletions and correlation with distinct neurological disease traits in females: further evidence for a contiguous gene syndrome. Hum Mutat. 2020;41(1):150–68.
Jin H, May M, Tranebjaerg L, Kendall E, Fontán G, Jackson J, Subramony SH, Arena F, Lubs H, Smith S, Stevenson R, Schwartz C, Vetrie D. A novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindness. Nat Genet. 1996;14(2):177–80.
Richter D, Conley ME, Rohrer J, Myers LA, Zahradka K, Kelecić J, Sertić J, Stavljenić-Rukavina A. A contiguous deletion syndrome of X-linked agammaglobulinemia and sensorineural deafness. Pediatr Allergy Immunol. 2001;12(2):107–11.
Pizzuti A, Fabbrini G, Salehi L, Vacca L, Inghilleri M, Dallapiccola B, Berardelli A. Focal dystonia caused by Mohr–Tranebjaerg syndrome with complete deletion of the DDP1 gene. Neurology. 2004;62(6):1021–2.
Sedivá A, Smith CI, Asplund AC, Hadac J, Janda A, Zeman J, Hansíková H, Dvoráková L, Mrázová L, Velbri S, Koehler C, Roesch K, Sullivan KE, Futatani T, Ochs HD. Contiguous X-chromosome deletion syndrome encompassing the BTK, TIMM8A, TAF7L, and DRP2 genes. J Clin Immunol. 2007;27(6):640–6.
Jyonouchi H, Geng L, Törüner GA, Vinekar K, Feng D, Fitzgerald-Bocarsly P. Monozygous twins with a microdeletion syndrome involving BTK, DDP1, and two other genes; evidence of intact dendritic cell development and TLR responses. Eur J Pediatr. 2008;167(3):317–21.
Brookes JT, Kanis AB, Tan LY, Tranebjaerg L, Vore A, Smith RJ. Cochlear implantation in deafness-dystonia-optic neuronopathy (DDON) syndrome. Int J Pediatr Otorhinolaryngol. 2008;72(1):121–6.
Arai T, Zhao M, Kanegane H, van Zelm MC, Futatani T, Yamada M, Ariga T, Ochs HD, Miyawaki T, Oh-ishi T. Genetic analysis of contiguous X-chromosome deletion syndrome encompassing the BTK and TIMM8A genes. J Hum Genet. 2011;56(8):577–82.
Shaker M, Lorigiano TH, Vadlamudi A. Xq22.1 contiguous gene deletion syndrome of X-linked agammaglobulinemia and Mohr–Tranebjærg syndrome. Ann Allergy Asthma Immunol. 2016;116(6):578–9.
Szaflarska A, Rutkowska-Zapała M, Gruca A, Szewczyk K, Bik-Multanowski M, Lenart M, Surman M, Kopyta I, Głuszkiewicz E, Machnikowska-Sokołowska M, Gruszczyńska K, Pituch-Noworolska A, Siedlar M. Neurodegenerative changes detected by neuroimaging in a patient with contiguous X-chromosome deletion syndrome encompassing BTK and TIMM8A genes. Cent Eur J Immunol. 2018;43(2):139–47.
Grillo L, Reitano S, Belfiore G, Spalletta A, Amata S, Bottitta M, Barone C, Falco M, Fichera M, Romano C. Familial 1.1 Mb deletion in chromosome Xq22.1 associated with mental retardation and behavioural disorders in female patients. Eur J Med Genet. 2010;53(2):113–6.
Shimojima K, Okanishi T, Yamamoto T. Marfanoid hypermobility caused by an 862 kb deletion of Xq22.3 in a patient with Sotos syndrome. Am J Med Genet A. 2011;155A(9):2293–7.
Labonne JD, Graves TD, Shen Y, Jones JR, Kong IK, Layman LC, Kim HG. A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies. BMC Neurol. 2016;16:132.
Cao Y, Aypar U. A novel Xq22.1 deletion in a male with multiple congenital abnormalities and respiratory failure. Eur J Med Genet. 2016;59(5):274–7.
Shirai K, Higashi Y, Shimojima K, Yamamoto T. An Xq22.1q22.2 nullisomy in a male patient with severe neurological impairment. Am J Med Genet A. 2017;173(4):1124–7.
Vaglio A, Greif G, Bernal M, Sanguinetti C, Mechoso B, Quadrelli A, Tucci P, Milunsky JM, Huang XL, Pagano S, Quadrelli R. Prenatal and postnatal characterization of a de novo Xq22.1 terminal deletion. Genet Test. 2006;10(4):272–6.
Wang H, Wang L, Yang J, Yin L, Lan L, Li J, Zhang Q, Wang D, Guan J, Wang Q. Phenotype prediction of Mohr–Tranebjaerg syndrome (MTS) by genetic analysis and initial auditory neuropathy. BMC Med Genet. 2019;20(1):11.
Swerdlow RH, Wooten GF. A novel deafness/dystonia peptide gene mutation that causes dystonia in female carriers of Mohr–Tranebjaerg syndrome. Ann Neurol. 2001;50(4):537–40.
Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007;9(1):34–45.
Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, Brühl K, Gal A, Bunge S, Beck M. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24(7):715–24.
Hurst S, Garbern J, Trepanier A, Gow A. Quantifying the carrier female phenotype in Pelizaeus–Merzbacher disease. Genet Med. 2006;8(6):371–8.
Acknowledgements
We appreciate the family for their contribution to this study.
Funding
This work was supported by grants from the Science and Technology Bureau of Taizhou (20ywa13, 20ywb05). The funding body did not have any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
HHX and WWS: concept, acquisition of data, article draft, revised article critically, corresponding author; YZ and ZHH: performed the karyotype analysis; WWS: provided a genetic counselling to the family and revised article critically; XHD and FYP: carried out CNV-seq experiments and analyzed obtained results; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Medical Ethics Review Board of Taizhou Hospital in Zhejiang Province, China (approval # K20200804). Written informed consent was obtained from the individual or guardian participants.
Consent for publication
Written informed consent was obtained from the individual or guardian participants for publication of this study, including their medical data and images.
Competing interests
The authors declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. G-banded karyotypes of the foetus and her parent.
Additional file 2
. ClinGen database shown 5.29 Mb deletion in chromosome Xq22.1–q22.3 affect 98 genes from DRP2 to NAP1L4P2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, HH., Zhang, Y., He, ZH. et al. Familial 5.29 Mb deletion in chromosome Xq22.1–q22.3 with a normal phenotype: a rare pedigree and literature review. BMC Med Genomics 16, 111 (2023). https://doi.org/10.1186/s12920-023-01547-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01547-2