
Abstract
Background
Although Chromosomal microarray analysis (CMA) is a powerful diagnostic technology for detecting chromosomal copy number variants (CNVs), it detects numerous variants of unknown significance (VUSs), which poses a great challenge for genetic counselling. Terminal deletion of the long arm of chromosome 4 is a rare genetic aberration. Few cases of interstitial deletion sharing the common deleted segment have been reported.
Case presentation
A male foetus with a 7.22-Mb deletion at chromosome 4q32.2q32.3 was found in the proband. The paternal genotype was normal. His asymptomatic mother with a normal phenotype and intelligence was found to carry the same deletion at the long arm of chromosome 4. The clinical significance of arr[GRCh37] 4q32.2q32.3(162858958_170081268)×1 remains uncertain. To the best of our knowledge, this is the first case report on a VUS of 4q32 deletion and the second report of a heterochromatic CNV involving part of the long arm of chromosome 4 in a phenotypically normal mother and child. The identification of this case contributes to additional understanding of deletion at 4q32.2q32.3. This report may provide a reference for prenatal diagnosis and genetic counselling in patients who have genotypes of similar cytogenetic abnormalities.
Conclusions
The novel 7.22-Mb deletion at chromosome 4q32.2q32.3 (162858958-170081268) is a VUS. The foetus inherited this VUS from a phenotypically normal mother.
Similar content being viewed by others


Background
Chromosomal microarray analysis (CMA) is currently widely used in prenatal diagnosis. It mainly has two technology platforms: array-based comparative genomic hybridization (aCGH) and single nucleotide polymorphism array (SNP array) [1]. Although CMA is a powerful diagnostic technology for detecting chromosomal copy number variants (CNVs) [2], it detects numerous variants of unknown significance (VUSs). These VUSs pose a great challenge for genetic counselling. Typically, a VUS is a rare or novel CNV in that is not known to have correlation with a clinical disease, and its pathogenicity can neither be ruled out nor confirmed. Based on the existing literature and databases, a VUS may be classified as “likely pathogenic”, “uncertain significance” or “likely benign” [3]. Parental verification testing can assist with classification because it can determine whether the variant occurred as a de novo or inherited genetic mutation in the foetus.
Terminal deletion of the long arm of chromosome 4 is a rare genetic aberration with an estimated prevalence of 1:100,000 [4]. Clinically, this kind of genetic aberration is known as “chromosome 4q-syndrome”. The common phenotypic features of 4q-syndrome include limb abnormalities, cardiac malformations, mental retardation, developmental delay, dysmorphic facial anomalies, Pierre Robin sequence and digital anomalies [5, 6]. In 1967, Ockey et al. first reported a deletion of the long arm of chromosome 4 in a child with limb abnormalities [7]. Since then, hundreds of patients with chromosome 4q-syndrome have been reported [4, 8,9,10,11]. The severity of the phenotype is correlated with the size of the deleted ranges (larger or smaller). It has been reported that del(4)(q32q33) has mild to moderate clinical symptoms [12]. Usually, deletions involving 4q31 determine more severe malformations than deletions involving band 4q34 [13].
In this case, we present a novel VUS of a 7.2-Mb deletion at chromosome 4q32.2q32.3 and illustrate the importance of reporting unusual variant chromosomes for genetic counselling purposes.
Case presentation
A 31-year-old healthy pregnant woman, gravida 2 para 1, underwent non-invasive prenatal testing (NIPT) at 15 weeks of gestation at Xingtai local hospital. NIPT was performed as previously described [14]. The result of NIPT indicated a 7.35 Mb deletion at chromosome 4q32.2q32.3(162582601-169932600del). There are several possible reasons for this result: chromosomal abnormalities in the mother, chromosomal abnormalities in the foetus, and chromosomal abnormalities in the placenta. The woman was then referred to our hospital at 22 weeks of gestation for prenatal diagnosis and genetic counselling. Her family and previous histories were uneventful. Her pregnancy history resulted in the term birth of a healthy boy. The sonographic examination did not reveal any ultrasound anomalies. After being informed about the possible risk, the woman decided to undergo amniocentesis. Prenatal karyotyping and CMA techniques were subsequently performed.
Methods and results
Chromosome analysis was carried out on cultured cells obtained from amniotic fluid by conventional Giemsa-band karyotyping at approximately 320-band resolution. The cytogenetic analysis revealed an apparently normal karyotype of 46, XY (Fig. 1) with limited banding resolution.

G-banded karyotype of the foetus indicated an apparently normal karyotype of 46, XY
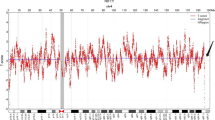
Microarray-based copy number analysis was performed using the Chromosome Analysis Suite software version 4.0.0.385 (R28959) (Thermo Fisher Scientific Inc.) and the results were presented on the International System for Human Cytogenomic Nomenclature 2016 (ISCN, 2016). To interpret the results of the SNP array, we used the Database of Cases with Heteromorphisms (http://ssmc-tl.com/HMs.html), Decipher Database (DECIPHER, http://decipher.sanger.ac.uk/), the Database of Genomic Variants (DGV, http://www.ebi.ac.uk/dgva/), Online Mendelian Inheritance in Man (OMIM, http://omim.org/), Clinical Genome Resource, (Clingen, https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/) and PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) to determine the clinical significance of CNVs. CMA using the Affymetrix CytoScan 750 K SNP microarray (Affymetrix CytoScan 750 K Array, Santa Clara, California), was performed on DNA extracted from amniotic fluid and a 7.22-Mb deletion was detected at chromosome 4q32.2q32.3 or arr[GRCh37] 4q32.2q32.3(162858958_170081268)×1 (Fig. 2). SNP array analyses of the parental blood showed that the paternal chromosomes were normal, but the maternal chromosomes had exactly the same deleted region as the foetus at chromosome 4q32.2q32.3 (Fig. 2).

SNP array analysis showing that the paternal chromosomes were normal, while the foetus and the mother had the same 7.22-Mb deletion at 4q32.2q32.3. (The color of purple represents the result of the foetus. The color of pink represents the result of the mother. The color of light blue represents the result of the father. The red block indicates the position of the 4q32.2q32.3 deletion.)
After genetic counselling, the couple decided to continue the pregnancy and a male neonate weighting 3050 g was born by caesarean section on November 12, 2019, with a five-minute Apgar score of 10 points, and no abnormal clinical symptoms or signs have been observed to date.
Discussion
In this case, the G-band karyotyping analysis of the foetus indicated an apparently normal karyotype of 46, XY (Fig. 1). As a gold standard procedure, conventional Giemsa-band karyotyping on metaphase cells can detect chromosomal aberrations at a resolution of 5–10 Mb [15]. Thus, deletions or duplications that are smaller than 5–10 Mb may be overlooked unless additional techniques are used [16]. CMA can provide much greater resolution and potentially detect CNVs and differences in the amount of chromosome samples as small as 100 to 200 kb [1]. The SNP array analysis of uncultured cells obtained from amniotic fluid showed a result of arr[GRCh37] 4q32.2q32.3(162858958_170081268)×1 (Fig. 2). This result is almost consistent with the previous NIPT result. Unexpectedly, no benign reports about this loss were found in the DGV database; no pathogenicity was reported in Decipher Database; and no dose sensitivity was reported in the Clingen database.
A total of 47 genes (including 15 OMIM genes) with already known or unknown functions have been mapped to the 7.22-Mb deleted region of chromosome 4q32.2q32.3(162858958-170081268). This region encompasses 3 protein coding and pathogenetic OMIM genes named Carboxypeptidase E (CPE) [MIM 114855], Tolloid Like 1 (TLL1) [MIM 606742] and Palladin, Cytoskeletal Associated Protein (PALD) [MIM 608092]. According to the Online Mendelian Inheritance in Man (OMIM, http://omim.org/), CPE is responsible for maturity-onset diabetes of the young, type 6 and hyperproinsulinaemia; TLL1 is involved in atrial septal defect 6 and atrial septal defect ostium primum; and PALLD is associated with pancreatic cancer 1 and pancreatic cancer. Other related genes in the 7.22-Mb deleted section may also contribute to the variable features, especially Methylsterol Monooxygenase 1 (MSMO1) [MIM 607545], which is known to be associated with diseases including congenital cataract, microcephaly and psoriasiform dermatitis.
In the existing literature, more than 100 cases have been previously reported on 4q deletions [4, 8,9,10,11]. However, no cases had interstitial deletions sharing the same deleted segment as the present case. To date, only a dozen cases involving the affected region of 4q32 have been reported (Table 1). In 1992, an 18-month-old girl with a cardiac defect, duplicated kidney, and postnatal growth failure was reported to have a distal deletion of 4q3l .22-q34.2, detected by using high resolution G banding on lymphocytes [17]. The genotype of del(4)(q32q34) is the most common 4q deletion and contributes to mild to moderate clinical symptoms including developmental delay [19,20,21] and congenital heart defects [23]. The most systematic research was performed by Strehle E M et al. [11]. They characterized 20 patients with 4q deletion syndrome by using array CGH and compared the clinical characteristics found in these patients with those of the 101 patients reviewed by Strehle and Bantock [4]. In addition, a 6-month-old boy presenting with congenital heart disease and clenched hands was reported with an interstitial deletion at 4q31-q32 [13]. More recently, Tidrenczel Z et al. [9] reported a prenatal diagnosis of a foetus with a 33.5 Mb deletion of 4q32.1-q35.2 presenting with high-risk combined screening test results and second trimester ultra-sound soft markers. Aladhami S M S et al. [18] reported a maternal inherited del(4)(q32q33) which were not leading to major malformations in affected persons. A 12-year-old proband and his 29-year-old mother both showed mild dysmorphic features, obesity, late presentation of learning difficulties and behavior problems [18]. This case lost the whole region of 4q32 and 4q33 (approximately 18 Mb large) which comprised the affected region of our case (7.22-Mb deletion at 4q32.2q32.3) completely. Generally, the smaller the deletions are the milder are the phenotypes as compared with larger deletions. Interestingly, a 22-year-old individual reported by Tzschach A et al. [22] with mild to moderate mental retardation, psychosis and obesity was found to have a 4q32.1-q32.3 deletion, which comprised a deleted region the most similar to our case but smaller than the case del(4)(q32q33) discussed above. According to Tzschach A et al. [22], the 10-Mb deletion at 4q32.1q32.3 was harbored more than 30 genes, and haploinsufficiency of one or several of these genes is likely to have caused the clinical problems of the patient. The above patients possessed distal 4q deletions that overlapped with our case, but no individuals with a reported interstitial deletion were identical to the deletion found in the proband.
Based on what has been discussed above, the 7.22-Mb deletion of chromosome 4q32.2q32.3 is a VUS. Its pathogenicity cannot be ruled out definitely. The VUS in prenatal diagnosis usually pose a great challenge for genetic counselling. The information obtained from prenatal diagnosis could facilitate prospective parents’ reproductive decision-making when confronted with the choice between terminating pregnancy and continuing pregnancy. How do we avoid ending a potentially benign life; but avoid the pain and suffering that a defective child may bring to a family? This is not easy to answer when encountering VUSs clinically. Most women wish to be reassured that their unborn child is healthy. Inevitably, any prenatal diagnosis programme that aims to provide such reassurance will cause anxiety, especially those with diagnoses of unknown clinical significance. Beneficial and clear counselling may ease the anxiety of a pregnant woman and reduce the chance of medical disputes.
To further clarify the clinical significance of the proband, parental verification tests are subsequently required. SNP array analyses of the parental blood showed that the paternal chromosomes were normal, while the maternal chromosomes had exactly the same deletion region at chromosome 4q32.2q32.3 compared with that of the foetus (Fig. 2). Usually, when a mother and her unborn baby carry the same VUS at an autosome, the baby is less likely to be pathogenic if the mother is healthy and normal. To the best of our knowledge, this is the first case report on a VUS of 4q32 deletion and the second report of a CNV involving part of the long arm of chromosome 4 in a phenotypically normal mother and child. Docherty Z et al. first reported a prenatal case of a rare heterochromatic variant on chromosome 4 in 1984. A phenotypically normal foetus inherited the abnormal karyotype of 46,XY,add(4)(q35)? from a clinically healthy woman [24].
Conclusion
The novel 7.22-Mb deletion at chromosome 4q32.2q32.3(162858958-170081268) is a VUS. The foetus inherited this variant from an asymptomatic and healthy pregnant woman without any ultrasound anomalies. It appears that the 7.22-Mb deletion is a rare heterochromatic variant. After genetic counselling, the couple decided to continue the pregnancy and a male neonate with a normal phenotype was born at 39 plus 3 weeks of pregnancy.
Availability of data and materials
NA
Abbreviations
- CMA:
-
Chromosomal microarray analysis
- SNP array:
-
Single nucleotide polymorphism array
- aCGH:
-
Array-based comparative genomic hybridization
- Mb:
-
Megabasepairs
- UPD:
-
Uniparental disomy
- CNVs:
-
Copy number variants
- NIPT:
-
Non-invasive prenatal testing
- VUSs:
-
Variants of unknown significance
- ISCN, 2016:
-
the International System for Human Cytogenomic Nomenclature 2016
References
Van den Veyver IB, Beaudet AL. Comparative genomic hybridization and prenatal diagnosis. Curr Opin Obstet Gynecol. 2006;18(2):185–91 https://doi.org/10.1097/01.gco.0000192986.22718.cc.
Phadke S. Cytogenetic microarray in prenatal and postnatal diagnosis. Mol Cytogenet. 2014;7(1):I32 https://doi.org/10.1186/1755-8166-7-S1-I32.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405 https://doi.org/10.1038/gim.2015.30.
Strehle EM, Bantock HM. The phenotype of patients with 4q-syndrome. Genet Couns. 2003;14(2):195–205 PMID: 12872814.
Strehle EM, Ahmed OA, Hameed M, et al. The 4q-syndrome. Genet Couns. 2001;12(4):327–39 PMID: 11837601.
Vona B, Nanda I, Neuner C, et al. Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: review of literature. BMC Med Genet. 2014;15:1–7 https://doi.org/10.1186/1471-2350-15-72.
Ockey CH, Feldman GV, Macaulay ME, et al. A large deletion of the long arm of chromosome No. 4 in a child with limb abnormalities. Arch Dis Child. 1967;42(224):428 https://doi.org/10.1136/adc.42.224.428.
Wu Y, Wang Y, Wen SW, et al. Recombinant chromosome 4 in two fetuses-case report and literature review. Mol Cytogenet. 2018;11(1):48 https://doi.org/10.1186/s13039-018-0393-1.
Tidrenczel Z, Tardy EP, Pikó H, et al. Prenatal diagnosis of 4q terminal deletion and review of the literature. Cytogenet Genome Res. 2019;158(2):63–73 https://doi.org/10.1159/000500735.
Vlaikou AM, Manolakos E, Noutsopoulos D, et al. An interstitial 4q31. 21q31. 22 microdeletion associated with developmental delay: case report and literature review. Cytogenet Genome Res. 2014;142(4):227–38 https://doi.org/10.1159/000361001.
Strehle EM, Yu L, Rosenfeld JA, et al. Genotype–phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet A. 2012;158(9):2139–51 https://doi.org/10.1002/ajmg.a.35502.
Liehr T. Cases with Heteromorphisms. 2019. http://ssmc-tl.com/HMs.html. Accessed 18 Dec 2019.
Ismail S, Helmy NA, Mahmoud WM, et al. Phenotypic characterization of rare interstitial deletion of chromosome 4. J Pediatr Genet. 2012;1(3):189–94 https://doi.org/10.3233/PGE-2012-029.
Zhang H, Gao Y, Jiang F, et al. Non-invasive prenatal testing for trisomies 21, 18 and 13: clinical experience from 146 958 pregnancies. Ultrasound Obstet Gynecol. 2015;45(5):530–8 https://doi.org/10.1002/uog.14792.
Beulen L, Faas BHW, Feenstra I, et al. Clinical utility of non-invasive prenatal testing in pregnancies with ultrasound anomalies. Ultrasound Obstet Gynecol. 2017;49(6):721–8 https://doi.org/10.1002/uog.17228.
Lichtenbelt KD, Knoers N, Schuring-Blom GH. From karyotyping to array-CGH in prenatal diagnosis. Cytogenet Genome Res. 2011;135(3–4):241–50 https://doi.org/10.1159/000334065.
Sarda P, Lefort G, Fryns JP, et al. Interstitial deletion of the distal long arm of chromosome 4. J Med Genet. 1992;29(4):259–61 https://doi.org/10.1136/jmg.29.4.259.
Aladhami SM, Gould CP, Muhammad FA. A new inherited interstitial deletion of the distal long arm of chromosome 4. Hum Hered. 2000;50(2):146–50 https://doi.org/10.1159/000022904.
Keeling SL, Lee-Jones L, Thompson P. Interstitial deletion 4q32-34 with ulnar deficiency: 4q33 may be the critical region in 4q terminal deletion syndrome. Am J Med Genet. 2001;99(2):94–8 https://doi.org/10.1002/1096-8628.
Ramanathan S, Woodroffe A, Flodman PL, et al. A case of autism with an interstitial deletion on 4q leading to hemizygosity for genes encoding for glutamine and glycine neurotransmitter receptor sub-units (AMPA 2, GLRA3, GLRB) and neuropeptide receptors NPY1R, NPY5R. BMC Med Genet. 2004;5(1):10 https://doi.org/10.1186/1471-2350-5-10.
Kaalund SS, Møller RS, Tészás A, et al. Investigation of 4q-deletion in two unrelated patients using array CGH. Am J Med Genet A. 2008;146(18):2431–4 https://doi.org/10.1002/ajmg.a.32458.
Tzschach A, Menzel C, Erdogan F, et al. Characterization of an interstitial 4q32 deletion in a patient with mental retardation and a complex chromosome rearrangement. Am J Med Genet A. 2010;152(4):1008–12 https://doi.org/10.1002/ajmg.a.33343.
Xu W, Ahmad A, Dagenais S, et al. Chromosome 4q deletion syndrome: narrowing the cardiovascular critical region to 4q32. 2-q34. 3. Am J Med Genet A. 2012;158(3):635–40 https://doi.org/10.1002/ajmg.a.34425.
Docherty Z, Bowser-Riley SM. A rare heterochromatic variant of chromosome 4. J Med Genet. 1984;21:470–2 https://doi.org/10.1002/ajmg.a.20594.
Acknowledgments
The authors would like to thank the proband’s family for the cooperation.
Funding
This study was supported by the project of Health commission of Hebei province (20180117 and 20190209).
Author information
Authors and Affiliations
Contributions
All authors contributed to this work equally. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All individuals have given their written consent to participate in this study.
Consent for publication
The authors have obtained written consent to publish patient images.
Competing interests
The authors declare that there are no conflicts of interest regarding the publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, P., Sun, Y., Huo, P. et al. Prenatal diagnosis of a maternal 7.22-Mb deletion at chromosome 4q32.2q32.3 by SNP array. Mol Cytogenet 13, 12 (2020). https://doi.org/10.1186/s13039-020-00480-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-020-00480-8