
Abstract
Background
Breast cancer (BRCA) is the most common malignancy with high morbidity and mortality in women, and transcription factor (TF) is closely related to the occurrence and development of BRCA. This study was designed to identify a prognostic gene signature based on TF family to reveal immune characteristics and prognostic survival of BRCA.
Methods
In this study, RNA-sequence with corresponding clinical data were obtained from The Cancer Genome Atlas (TCGA) and GSE42568. Prognostic differentially expressed transcription factor family genes (TFDEGs) were screened to construct a risk score model, after which BRCA patients were stratified into low-risk and high-risk groups based on their corresponding risk scores. Kaplan–Meier (KM) analysis was applied to evaluate the prognostic implication of risk score model, and a nomogram model was developed and validated with the TCGA and GSE20685. Furthermore, the GSEA revealed pathological processes and signaling pathways enriched in the low-risk and high-risk groups. Finally, analyses regarding levels of immune infiltration, immune checkpoints and chemotactic factors were all completed to investigate the correlation between the risk score and tumor immune microenvironment (TIME).
Results
A prognostic 9-gene signature based on TFDEGs was selected to establish a risk score model. According to KM analyses, high-risk group witnessed a significantly worse overall survival (OS) than low-risk group in both TCGA-BRCA and GSE20685. Furthermore, the nomogram model proved great possibility in predicting the OS of BRCA patients. As indicted in GSEA analysis, tumor-associated pathological processes and pathways were relatively enriched in high-risk group, and the risk score was negatively correlated with ESTIMATE score, infiltration levels of CD4+ and CD8+T cells, as well as expression levels of immune checkpoints and chemotactic factors.
Conclusions
The prognostic model based on TFDEGs could distinguish as a novel biomarker for predicting prognosis of BRCA patients; in addition, it may also be utilized to identify potential benefit population from immunotherapy in different TIME and predict potential drug targets.
Similar content being viewed by others


Introduction
To date, breast cancer (BRCA) has been the most common cancer among women worldwide, with high morbidity and mortality [1]. Over the last several decades, significant progress has been made in the field of BRCA treatment, including surgery, radiotherapy, chemotherapy, hormonal therapy, targeted therapy, immunotherapy and so on [2,3,4,5,6,7]. Nevertheless, the mortality for BRCA remains high due to its complicated pathogenesis, development and metastasis [8].
As is known to all, transcription factors (TFs) play an important role in promoting or inhibiting downstream genes via binding specific sequences, with significant influence on the occurrence, migration, invasion and other biological processes of tumors [9]. Meanwhile, TFs are reported to be crucial to the occurrence and development of BRCA [10, 11]. Recently, the role of tumor immune microenvironment (TIME) in BRCA and its effect on tumor progression have increasingly aroused general concern [12, 13]. It is worth noting that, TFs can also affect the prognosis of BRCA by various TIME, with specific mechanism remaining unclear [14, 15]. Therefore, it is urgently required to explore the underlying molecular mechanisms and prognostic indicators for patients with BRCA.
In some previous studies, bioinformatics analysis was completed based on prognosis model of malignant tumors in terms of lncRNA, circRNA or mRNA [16,17,18,19]. In our study, we acquired RNA-sequence with corresponding clinicopathological data of BRCA patients from The Cancer Genome Atlas (TCGA) and GSE42568, after which we conducted a comprehensive bioinformatics analysis based on levels of gene-expression, Venn analysis and univariate Cox analysis with clinical data from TCGA-BRCA. By selecting prognostic differentially expressed transcription factor family genes (TFDEGs), we managed to set up a risk score system of BRCA for validation in cohorts of TCGA and GSE20685. To further investigate the potential mechanisms and pathways thereof, the function and gene set enrichment analyses (GSEA) were completed on the basis of differentially expressed genes (DEGs) from high-risk group compared with low-risk group; meanwhile, Connectivity Map (CMap) database was used to predict potential drug targets for high-risk group. In addition, various analyses with regard to tumor-infiltrating immune cells (TIICs), immune checkpoints and chemotactic factors were adopted to clarify the correlation between the risk score and TIME.
Materials and methods
Data sources
RNA-sequence with corresponding clinical data of BRCA and normal samples were obtained from TCGA (https://portal.gdc.cancer.gov/) [20], consisting of 1109 BRCA patients with comprehensive profiles of gene expression and clinical characteristics, 40 of which were removed for incomplete transcriptomic and clinical information. After that, the remaining 1069 BRCA patients with complete information were included as training set for further analyses. The GSE42568 set, including 104 BRCA samples and 17 normal samples, was downloaded from Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.go/geo/), while TF family including 1536 genes were retrieved from the Molecular Signatures Database (MSigDB) (www.gsea-msigdb.org/gsea/msigdb/human/gene_familes.jsp). In addition, the cohort of GSE20685 (involving 327 BRCA samples) in GEO (https://www.ncbi.nlm.nih.go/geo/) was regarded as the testing set, including RNA-sequencing configuration files and clinical information.
Selection and functional clustering analyses of TFDEGs
The DEGs between tumor and normal samples were identified using the “DESeq2” package in TCGA-BRCA and “GEO2R” in GSE42568 with thresholds of |log2 (fold-change)|values > 1 and adjusted P < 0.05. Meanwhile, Venn analysis was applied to select overlapping DEGs among three algorithms mentioned above, and the “ggplot2” package to generate Volcano Plot and Differential Ranking Chart of the TFDEGs. By means of “clusterProfiler” package (version 3.14.3), Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses (www.kegg.jp/kegg/kegg1.html) were performed for patients based on the 113 TFDEGs [21]. Moreover, a protein–protein interaction (PPI) network of 113 TFDEGs was generated via Search Tool for the Retrieval of Interacting Genes (STRING) (https://string-db.org).
Constructing and validating risk score model
Based on clinical data of TCGA-BRCA, univariate Cox analysis of overall survival (OS) was applied to identify TFDEGs with prognostic values and then visualized by forest plot (P < 0.05) [22]. Least Absolute Shrinkage and Selection Operator (LASSO) regression model was conducted to reduce the overfitting high-dimensional prognostic genes [23]. Then, the screened genes were integrated into a risk signature, after which a risk score system was established based on the normalized values of gene expression and coefficients in accordance with the following formula.
The risk score was calculated for each BRCA patient, after which patients were divided into low-risk and high-risk groups according to the median risk score of TCGA-BRCA. Furthermore, patients were also assigned into subgroups by clinicopathological characteristics [including age, T stage, N stage, M stage, TNM stage, Menopause status (Pre: < 6 months since last menstrual period AND no prior bilateral ovariectomy AND not on estrogen replacement; Post: prior bilateral ovariectomy OR > 12 months since last menstrual period with no prior hysterectomy), and PAM50], so as to identify the correlation between risk score and clinical features. In addition, Kaplan–Meier (KM) analysis was utilized to evaluate the relationship between risk groups and OS of different groups.
Nomogram
To assess whether the risk score model could serve as an independent predictive indicator, univariate and multivariate Cox regression analyses were completed with clinicopathological indicators, including the age, T stage, N stage, M stage, status of estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2), as well as menopause. All the independent prognostic clinicopathological parameters and risk scores were summarized to establish a nomogram for predicting the OS rates of 2-, 3- and 5-year. Furthermore, calibration, time-dependent receiver operating characteristic (ROC) curves and concordance index (C-index) were applied to assess the discriminatory ability of the nomogram.
Analyses of 9 TFDEGs in risk score model
We performed differential expression analysis of identified risk genes in TCGA-BRCA, among which GO and KEGG pathway enrichment analyses with P values < 0.05 were regarded as statistically significant. Furthermore, Human Protein Atlas (HPA) database (http://www.proteinatlas.org/) was applied to evaluate the protein expression of the risk genes.
DNA methylation plays a key role in prognostic assessment and potential biomarker in cancer development [24]. MethSurv (https://biit.cs.ut.ee/methsurv/) was adopted in this study to determine the expression and prognostic patterns of single CpG methylation of the TFDEGs in BRCA [25]. In this analysis, DNA methylation values were represented by beta values ranging from 0 to 1.
Analysis of DEGs from high-risk group
DEGs from high-risk group compared with low-risk group in TCGA-BRCA were screened by R packages mentioned above, after which GO and KEGG pathway enrichment analyses with P < 0.05 were considered statistically significant. In addition, GSEA (http://software.broadinstitute.org/gsea/index.jsp) (version 3.14.3) [26] was applied to identify hallmarks of high-risk group, comparing with low-risk group, and get visualized by ridge map. Finally, a signature of DEGs from high-risk group compared with low-risk group was used to predict potential drug targets for BRCA patients in high-risk group through CMap database (https://clue.io) [27].
The correlation between prognostic model and TIME
As is well-known to all, the tumor infiltrates immune cells resulting in great threaten to patients' survival. The immune infiltration scores of TCGA-BRCA samples were computed by ESTIMATE [28], and the infiltration levels of 24 types of immune cells in BRCA samples were calculated through Immune Cell Abundance Identifier (ImmuCellAI) (http://bioinfo.life.hust.edu.cn/ImmuCellAI) [29]. In addition, we also validated the correlation between identified genes (TFDEGs) and immune cells by means of the "TIMER" (http://timer.cistrome.org/) analysis tool [30]. To further predict the TIME, the correlations of prognostic model with the expression of immune checkpoints and chemotactic factors were analyzed based on TCGA-BRCA.
Statistical analysis
Statistical analyses in this study were performed via R software (version 3.6.3) (R code, data input, and output of this study were provided in Additional file 1: R code and data), with log-rank test for the KM analysis. Furthermore, T test or Wilcoxon test was adopted to evaluate the differences in the risk score among various clinical characteristic subtypes, as well as those differences in the level of immune infiltration, immune checkpoints and chemotactic factors between the low-risk and high-risk groups. *P < 0.05, **P < 0.01, ***P < 0.001.
Results
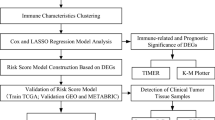
The detailed flowchart was shown in Fig. 1.

Flow diagram of the study
Identification of TFDEGs in BRCA patients
The DEGs were collected from sets of TCGA-BRCA (Fig. 2A) and GSE42568 (Fig. 2B). According to the criteria, we obtained 5073 DEGs from TCGA-BRCA and 3195 DEGs from GSE42568, and downloaded 1536 genes of TF family on the basis of MSigDB (Additional file 2: Table S1). Based on overlapping DEGs among the aforementioned three algorithms, a total of 113 TFDEGs were screened for further analysis (Fig. 2C).

Screening of TFDEGs in BRCA. Volcano plots of differentially expressed genes (DEGs) analysis in set of A TCGA-BRCA and B GSE42568 (the red and blue dots are up- and down-regulated genes); C Venn analysis indicating the overlap of genes among DEGs in TCGA-BRCA, GSE42568 set and transcription factor (TF) family genes from Molecular Signatures Database (MSigDB)
Expression and functional clustering analysis of TFDEGs in BRCA
Volcano Plot (Fig. 3A) and Differential Ranking Chart (Fig. 3B) of the 113 TFDEGs (75 up-regulated and 38 down-regulated) were hereby performed based on TCGA-BRCA, with Go and KEGG pathway enrichment analyses to reveal the functions of the 113 TFDEGs. Moreover, these genes were obviously enriched in terms of epithelial cell proliferation, DNA-binding transcription, transcription factor complex, and transcriptional mis-regulation in cancer (Fig. 3C), while the PPI network among the 113 TFDEGs also be conducted (Fig. 3D).

Expression and functional clustering analyses of 113 TFDEGs in BRCA. TFDEGs in TCGA-BRCA set displayed by: A Volcano Plot (the red and blue dots are up- and down-regulated genes) and B Differential Ranking Chart; C Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (www.kegg.jp/kegg/kegg1.html) analyses of TFDEGs; D protein–protein interaction (PPI) networks constructed by STRING
Constructing and validating risk score model
In this study, univariate Cox regression analysis was completed to explore the relationship between TFDEGs expression levels and OS in TCGA-BRCA. Eleven genes were identified as potential risk factors related to OS (Fig. 4A) by cut-off threshold of Cox P < 0.05, with the results of KM analyses shown in Additional file 3: Fig. S1. Furthermore, the LASSO regression algorithm was applied to refine the gene sets by calculating regression coefficients (Fig. 4B, C), after which 9 TFDEGs distinguished with most predictive value to establish the risk score model (Table 1). Based on the TFDEGs from the risk score model, the performance of ROC was then analyzed (Fig. 4D).

Construction and assessment of risk score model. A The 11 prognostic TFDEGs extracted by univariate Cox regression analysis and represented by a forest plot; B the tenfold cross-validation for variable selection in the LASSO model; C the LASSO coefficient profile of 11 TFDEGs; D receiver operating characteristic (ROC) curve suggesting the predictive precision of the risk score model; risk plot distribution, survival status of patients, and heat map including 9 TFDEGs in E TCGA-BRCA and F GSE20685; Kaplan–Meier (KM) survival curves of overall survival (OS) for patients between low-risk and high-risk groups in G TCGA-BRCA and H GSE20685
The risk score for each patient in TCGA-BRCA (training set) and GSE20685 (testing set) was calculated in accordance with the expression levels of 9 genes and their regression coefficients, as shown in Fig. 4E and F respectively. After that, patients were divided into low-risk and high-risk groups by median risk score. According to general distribution of survival times, patients with higher risk score might bear worse OS (Fig. 4E, F). Furthermore, the expression levels of the screened genes were investigated in this study (Fig. 4E, F). As indicated in KM analyses, high-risk group witnessed a significantly poorer clinical outcome than those of low-risk group in both training (P < 0.001, Fig. 4G) and testing sets (P = 0.003, Fig. 4H), attesting to our risk score model’s great potential for predicting the prognosis of BRCA patients.
As shown in Fig. 5A–G, the relationship between risk score and clinical characteristics in the training set (TCGA-BRCA) was analyzed in this study, suggesting obviously higher risk score in groups of patients with age > 60 years (P = 0.046), T stage: II (P < 0.001), TNM stage: II (P = 0.008), HER2 (compared with LumA or LumB: P = 0.005; compared with Basal: P = 0.020). Nevertheless, no obvious difference was found between the risk scores and N stage, M stage as well as menopause status.

The predictive power of the risk score system. Correlation between the risk score and clinical characteristics: A age; B T stage; C N stage; D M stage; E TNM stage; F menopause status; G PAM50; KM curves for OS prediction of in subgroups of H age ≤ 60 years; I age > 60 years; J T1&2; K T3&4; L N0&1; M N2&3; N M0; O LumA or LumB; P stages I&II; Q stages III&IV; R menopause status (pre) and S menopause status (post)
In our study, the prediction efficiency of risk groups was further validated by several subgroups. According to the KM analyses, high-risk patients generally had a worse OS in subgroups of age ≤ 60 years, age > 60 years, T1&2, T3&4, N0&1, N2&3, M0, LumA or LumB, Stages I&II, Stages III&IV, menopause status (pre) and menopause status (post) (Fig. 5H–S). However, no significant difference was found in subgroups of M1 and Her2 & Basal (Additional file 4: Fig. S2).
Nomogram
To determine whether the risk score model could be regarded as an independent risk factor for OS of BRCA patients, the potential prognostic indicators (age, T stage, N stage, M stage, ER status, PR status, HER2 status, and menopause status) were therefore analyzed via univariate and multivariate Cox regression in TCGA-BRCA (Table 2). For the purpose of in-depth study, all the independent prognostic clinicopathological parameters and risk scores were evaluated by univariate and multivariate Cox regression in TCGA-BRCA (Fig. 6A, B) and GSE20685 (Fig. 6C, D), indicating that high-risk group witnessed significantly worse OS in both training set [hazard ratio (HR) = 2.733, 95% confidence interval (95% CI) 1.705–4.379, P < 0.001] and testing set (HR = 3.670, 95% CI 1.592–8.460, P = 0.002). Therefore, the age, N stage and risk score were identified as independent risk factors for OS then integrated into nomogram model (Fig. 6E), with C-index (with corresponding 95% CI) of 0.712 (95% CI 0.685–0.739). After that, we calculated the score for each BRCA according, and the predictive capability and consistency of the nomogram were assessed by calibration curve. The calibration plots showed outstanding consistency among the 2-, 3-, and 5-year OS rates when comparing with the ideal and nomogram model in both training (Fig. 6F) and testing (Fig. 6G) sets. Besides, time-dependent ROC analysis was also utilized to assess the discriminatory ability of the nomogram (Fig. 6H, I).

Nomogram and verification of prognostic model. A univariate and B multivariate Cox analyses of clinical factors and risk score with OS in TCGA-BRCA; C univariate and D multivariate Cox regression analyses of clinical factors and risk score with OS in GSE20685; E nomogram predicting 2-, 3- and 5-years survival rate of BRCA patients; the calibration curves for 2-, 3-, and 5-year OS in F TCGA-BRCA and G GSE20685; time-dependent ROC curves for predictive performance of the model in H TCGA-BRCA and I GSE20685
The DEGs between groups and potential drug targets for high-risk group
To clarify the potential influence of the screened TFDEGs expression levels on transcriptomic profile of BRCA, Go and KEGG pathway enrichment analyses were performed in order to investigate the functions of DEGs from high-risk group, comparing with low-risk group in TCGA-BRCA (Fig. 7A), suggesting these genes significantly enriched in terms of T cell activation, cell chemotaxis, receptor ligand activity together with ion channel activity. Based on GSEA analysis of DEGs from high-risk group compared with low-risk group, pathways as those related to cell cycle, cellular senescence and DNA methylation were enriched in high-risk group, while T cell receptor signaling pathway, metabolism of lipids, chemokine receptors bind chemokines, and immunoregulatory interactions were enriched in low-risk group (Fig. 7B), indicating TFDEGs' potential role in the metabolism, progression and tumor microenvironment of BRCA.

The DEGs between groups and potential drug targets for high-risk group. A GO and KEGG analyses of DEGs from high-risk group compared with low-risk group; B ridge map of gene set enrichment analysis (GSEA) for DEGs from high-risk group compared with low-risk group; C Connectivity Map (CMap) analysis to find the potential drug targets for high-risk group. The 20 top and bottom drugs represented positive and negative correlations, respectively, with BRCA patients in high-risk group. CMap analysis identified terconazole, QX-314, and several histamine receptor modulators as potential therapeutic drugs for BRCA patients in high-risk group
Besides, we uploaded both up- and down-regulated DEGs into the CMap database to predict potential drug the for high-risk group. CMap applies a systematic approach to reveal interactions among drugs, compounds, and diseases based on DEGs from high-risk group compared with low-risk group. The top 20 drugs with positive correlations and the top 20 drugs with negative correlations were obtained from CMap (Fig. 7C). These drugs were ranked by p-values and were determined based on the DEGs signatures against the CMap database. Terconazole, QX-314, and several histamine receptor modulators may serve as potential therapeutic drugs for BRCA patients in high-risk group.
Analyses of 9 TFDEGs in risk score model
The 9 TFDEGs were significantly differential expression (4 up-regulated and 5 down-regulated) in TCGA-BRCA (Fig. 8A, B) and closely related to each other (Fig. 8C). In addition, immunohistochemical analysis in HPA was applied to further investigate the expression level of the 9TFDEGs (Fig. 8D).

Analyses of 9 TFDEGs in prognostic model. Expression of 9 TFDEGs between normal and tumor in TCGA-BRCA by A unpaired samples and B paired samples; C relationship among the 9 TFDEGs; D the protein expression of 9 TFDEGs in BRCA tissue by immunohistochemistry from HPA database
DNA methyltransferases on CpG island methylation are transcription factors in the suppression or promotion of cell growth which is a reversible process [31]. In this study, we present the heatmap and prognostic value of DNA methylation clustering the expression levels of the 9 TFDEGs in BRCA (Additional file 5: Fig. S3 and Additional file 6: Table S2). With regard to DNA methylation expression levels, cg01024618, cg05008688, cg07787851 from ZNF106; cg16233472, cg19694099, cg15453482 from OVOL1; cg22228071, cg18045515, cg22892904, cg07335357, cg14726117, cg17346145 from CBX2; cg19109431 from NFKBIE; cg12271317, cg00337658, cg11113607 from LEF1; cg07275757, cg10590842 from NR3C2 came up with the highest levels and significant prognostic values (likelihood ratio (LR) test P < 0.05) in BRCA.
Correlation between prognostic model and TIME
Based on this study, the risk score was negatively correlated with immune infiltration score (including Stromal score, Immune score, and ESTIMATE score) (Fig. 9A, B).

Correlation between prognostic model and TIME. A Relationship between immune infiltration scores (including Stromal score, Immune score, and ESTIMATE score) and risk score; B comparison of immune infiltration scores (including Stromal score, Immune score, and ESTIMATE score) between low-risk and high-risk groups; C correlations between risk model and tumor-infiltrating immune cells (TIICs); D comparisons of TIICs between low-risk and high-risk groups; E association between risk score and immune checkpoints; F comparison of six immune checkpoints between low-risk and high-risk groups; G association between risk score and chemotactic factors; H comparison of six chemotactic factors between low-risk and high-risk groups
In addition, the correlation between the prognostic model and immune cells infiltration of patients in TCGA-BRCA cohort were also taken into account, with the proportion of different infiltrating immune cells retrieved from ImmuCellAI. The results indicated that the risk score was positively correlated with neutrophil, monocyte, nTreg, Tem, DC, and Th17, while negatively correlated with Tfh, NK, CD4 + T cells, Tgd, CD8 + T cells, Tc, and NKT (Fig. 9C). Furthermore, the high-risk group generally witnessed less infiltrating immune cells, especially CD4 + and CD8 + T cells (Fig. 9D).
As analyzed with the TIMER tool, expressions of TFDEGs were also correlated with immune infiltration profiles in BRCA. In summary, expression of each TFDEGs gene was associated with tumor purity and markers of different immune cells (Additional file 7: Fig. S4).
Moreover, we evaluated the relationship between risk score model and immune checkpoints (Fig. 9E), to find the expression levels of PDCD1 (PD-1), CD274 (PD-L1), CTLA4, LAG3, TIGIT and CD96 negatively correlated with the risk score (Fig. 9F).
Finally, we analyzed the relationship between risk score model and chemotactic factors (Fig. 9G), suggesting that the expression levels of CCL2, CCR4, CXCR4, CCR2, CCR5 and CXCR2 were negatively correlated with the risk score (Fig. 9H).
Discussion
To our knowledge, BRCA lists as the leading cause of cancer-related mortality among women all over the world. Despite of remarkable progress in diagnosis and treatment of BRCA over the last decades, a wide array of problems regarding its progression, metastasis and treatment resistance are yet to be fully clarified [32,33,34].
TFs recognize specific DNA sequences to control chromatin and transcription, forming a complex system to guide the genome expression [9]. With a key role in human physiology, disease and variation, TFs are closely related to the occurrence and development of tumors [35,36,37].
In recent years, an increasing number of studies have found several TFs to participate in tumorigenesis, progression together with the microenvironment of BRCA, indicating their potential and promising role to serve as prognostic markers for BRCA [38,39,40,41]. Nevertheless, they appeared to be inconsistent due to small datasets, the heterogeneity of BRCA, as well as variation in data pre-processing approaches. It is worth noting that several studies have figured out multigene panels' possible role as prognostic indicators in BRCA [42,43,44,45,46], while all these studies only focused on the identification of prognostic signatures. Hence, this study targeted at a comprehensive profiling of TF family in BRCA patients, in order to develop a prognostic model and explore its correlation with TIME.
In this study, we obtained gene expression and clinicopathological data of BRCA from TCGA and GEO database, after which, by means of univariate Cox analysis and LASSO regression analysis, a risk score model composed of nine genetic biomarkers was therefore established based on TFDEGs. Furthermore, the risk score was verified to be both effective and stable for predicting the prognosis of low-risk and high-risk groups in TCGA-BRCA and subgroups via KM curve. In order to predict the OS of BRCA patients, we also established a prognostic nomogram model based on TFDEGs, with a comprehensive integration of the risk score, age and N stage. Calibration plots revealed the robust predictive ability of the prognostic nomogram for OS in TCGA and GSE20685 cohorts, suggesting our prognostic model's great potential in predicting the clinical outcomes of BRCA patients.
The 9-TFDEGs prognostic model was comprised of ZNF106, OVOL1, CBX2, NFKBIE, LEF1, KLF15, EGR3, NR3C2 and MEOX1, all of which were protein coding gene. In BRCA, ZNF106, KLF15, EGR3, NR3C2 and MEOX1 were significantly down-regulated, while OVOL1, CBX2, NFKBIE, LEF1 were obviously up-regulated.
With a variety of cellular functions, including insulin receptor signaling, rRNA transcriptional regulation, ZNF106 is essential for maintaining motor and sensory neurons [47]. According to Guo et al. [48] ZNF106 enjoyed great prognostic significance in tumors with considerably lower expression. OVOL1, a key mediator of epithelial lineage determination and mesenchymal–epithelial transition (MET), has been proved to be inversely correlated with the epithelial-mesenchymal transition (EMT) signature and serve as a great prognostic indicator for BRCA patients [49]. In addition, CBX2 encodes a component of the multiprotein complex, which is required to maintain the transcriptionally repressive state of many genes throughout development via chromatin remodeling and modification of histones. Furthermore, Iqbal et al. [50] have reported that CBX2 and CBX7 could predict the outcomes and sensitivity to FDA-approved/investigational drugs in BRCA, and pathways related to NFKBIE could lead to the activation of NF-KappaB by PKR and bacterial infections in CF airways. Study had revealed that it might be associated with the potential target of triple-negative breast cancer (TNBC) [51]. Moreover, the protein encoded by LEF1 can bind to a functionally important site in the T-cell receptor-alpha enhancer, thereby conferring maximal enhancer activity. As a TF of EMT, it is involved in the Wnt signaling pathway. Apart from EMT, LEF1 facilitates metastasis by improving the antioxidative capacity of epithelial BRCA cells [52]. As a putative BCCA suppressor gene, KLF15 was recorded to get involved in negative regulation of peptidyl-lysine acetylation, together with positive regulation of transcription by RNA polymerase II [53]. The protein of EGR3 encoded by this gene participates in the transcriptional regulation of genes in controlling biological rhythm, and it may also play a certain role in various processes including muscle development, lymphocyte development, endothelial cell growth and migration, as well as neuronal development. Furthermore, Inoue has found EGR3 involved in the estrogen-signaling pathway and closely associated with the genesis and malignant progression of BRCA [54]. The NR3C2 protein functions as a ligand-dependent TF binding to mineralocorticoid response elements in order to transactivate target genes. As reported, miR-301b may be a tumor-promoting miRNA in BRCA, and miR-301b/NR3C2 axis mediated tumor development from cell proliferation and migration [55]. Additionally, study had revealed that combined p53- and PTEN-deficiency in TNBC activated expression of the MEOX1, which might serve as a potential therapeutic target for managing p53- and PTEN-deficient TNBC [56].
Moreover, we performed GO and KEGG functional analyses based on DEGs between low-risk and high-risk groups, indicating that DEGs were significantly enriched in terms of T cell activation, cell chemotaxis, receptor ligand activity and ion channel activity, which were closely related to the formation and development of tumors and immune microenvironment. Therefore, we further conducted GSEA between low-risk and high-risk groups, suggesting that pathways associated with formation and progression of tumor were enriched in the high-risk group, while pathways related to tumor associated immune cells and immunotherapeutic responses regulation were enriched in the low-risk group.
In addition, through CMap analysis, we revealed that certain drugs with highly negative correlations might serve as a potential treatment for high-risk group. Of course, this result needs to be further verified in vitro and vivo. Terconazole (TCZ), an azole antifungal drug, has been shown to enhance the cytotoxicity of antimitotic drugs in P-glycoprotein-overexpressing-resistant cancer cells [57]. Fuseya et al. [58] have found that systemic administration of QX-314 in mice could inhibit some behavioral aspects of bone cancer pain through selective inhibition of TRPV1-expressing afferents without coadministration of TRPV1 agonists. Cell proliferation is critical for tumor development and progression, and histamine is a main mediator of this biological process in different types of cancers. It has been recorded that histamine receptor modulator could regulate cancer-associated biological processes during cancer development in multiple cell types, including neoplastic cells and cells in the tumor micro-environment [59]. Collectively, the results suggested that these drugs are potential therapeutic drugs for high-risk group.
Several studies have figured out that TIME was correlated with the prognosis of cancer patients [60,61,62,63]. To further clarify the relationship between TIME and the prognostic model, analysis was hereby performed to reveal the correlation between risk score and ESTIMATE score, tumor infiltrating immune cells, immune checkpoints as well as chemotactic factors.
In immune infiltration analyses, the risk score was found to be negatively correlated with ESTIMATE score, composing of Stromal score and Immune score. With regard to tumor infiltrating immune cells analysis, the risk score was negatively correlated with infiltration levels of CD4+ and CD8+T cells, which played a key role in immunotherapy [64, 65]. Based on immune checkpoints and chemotactic factors analyses, the established risk score was negatively correlated with the expression levels of immune checkpoints (such as PD1, PD–L1, CTLA4, LAG3, TIGIT, etc.) and chemotactic factors (including CCL2, CCR4, CXCR4, CCR2, CCR5, etc.). In summary, compared with BRCA in low-risk groups, tumors of high-risk groups tended to be immunologically “cold” and might not benefit from immunotherapy.
In conclusion, the prognostic model based on TFDEGs can well predict the prognosis of BRCA patients; moreover, it may also be utilized to screen the appropriate immunotherapy benefit population and predict potential drug targets. The top of agenda for future research is to precisely identify high-risk groups, reverse the occurrence and development of tumor by screening effective drug targets, and transform immunologically “cold” tumors into responsive “hot” lesions by changing the TIME (Fig. 10) [66,67,68].

A model for prognostic and immune signature of TFDEGs in BRCA
Conclusions
The prognostic model based on TFDEGs could distinguish as a novel biomarker for predicting prognosis of BRCA patients; in addition, it may also be utilized to identify potential benefit population from immunotherapy in different TIME and predict potential drug targets.
Availability of data and material
The data sets used and/or analyzed in this study are available from the cancer genome database (TCGA-BRCA) (https://portal.gdc.cancer.gov), NCBI Gene Expression Omnibus (GEO: datasets of GSE42568 and GSE20685) (https://www.ncbi.nlm.nih.gov/geo/), Molecular Signatures Database (MSigDB) (http://www.gsea-msigdb.org/gsea/msigdb/index.jsp), KEGG pathway database (www.kegg.jp/kegg/kegg1.html), Connectivity Map (CMap) database (https://clue.io), Human Protein Atlas (HPA) database (http://www.proteinatlas.org/): HPA054267 (patient id: 4193), HPA003984 (patient id: 2091), HPA023083 (patient id: 2805), HPA005941 (patient id: 1939), CAB019405 (patient id: 2898), HPA028866 (patient id: 2091), HPA006206 (patient id: 2428), CAB009155 (patient id: 2015), HPA045214 (patient id: 2160), MethSurv database (https://biit.cs.ut.ee/methsurv/), and ImmuCellAI (http://bioinfo.life.hust.edu.cn/ImmuCellAI) database. Besides, R code, data input, and output of this study were provided in Additional file 1: R code and data.
Code availability
Not applicable.
Abbreviations
- TFDEGs:
-
Differentially expressed transcription factor family genes
- DEGs:
-
Differentially expressed genes
- TF:
-
Transcription factor
- MSigDB:
-
Molecular Signatures Database
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein–protein interaction
- KM:
-
Kaplan–Meier
- GSEA:
-
Gene set enrichment analysis
- BRCA:
-
Breast cancer
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- OS:
-
Overall survival
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- ROC:
-
Receiver operating characteristic
- HPA:
-
Human Protein Atlas
- C-index:
-
Concordance index
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hazard ratio
- CIs:
-
Confidence intervals
- TNBC:
-
Triple-negative breast cancer
- TIME:
-
Tumor immune microenvironment
- TIICs:
-
Tumor-infiltrating immune cells
- CMap:
-
Connectivity map
- ImmuCellAI:
-
Immune Cell Abundance Identifier
- TIMER:
-
Tumor Immune Estimation Resource
- LR:
-
Likelihood ratio
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Matsen CB, Neumayer LA. Breast cancer: a review for the general surgeon. JAMA Surg. 2013;148(10):971–9.
Vaidya JS, Bulsara M, Wenz F, Tobias JS, Joseph D, Baum M. Targeted radiotherapy for early breast cancer. Lancet. 2018;391(10115):26–7.
Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389(10087):2430–42.
Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet. 2017;389(10087):2403–14.
Lancet T. Breast cancer targeted therapy: successes and challenges. Lancet. 2017;389(10087):2350.
Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24(3):511–20.
Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569(7757):560–4.
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;172(4):650–65.
Addison JB, Voronkova MA, Fugett JH, Lin CC, Linville NC, Trinh B, et al. Functional hierarchy and cooperation of EMT master transcription factors in breast cancer metastasis. Mol Cancer Res. 2021;19(5):784–98.
Hornsveld M, Smits LMM, Meerlo M, van Amersfoort M, Groot Koerkamp MJA, van Leenen D, et al. FOXO Transcription factors both suppress and support breast cancer progression. Cancer Res. 2018;78(9):2356–69.
Mehraj U, Dar AH, Wani NA, Mir MA. Tumor microenvironment promotes breast cancer chemoresistance. Cancer Chemother Pharmacol. 2021;87(2):147–58.
Mittal S, Brown NJ, Holen I. The breast tumor microenvironment: role in cancer development, progression and response to therapy. Expert Rev Mol Diagn. 2018;18(3):227–43.
Wei X, Chen L, Yang A, Lv Z, Xiong M, Shan C. ADRB2 is a potential protective gene in breast cancer by regulating tumor immune microenvironment. Transl Cancer Res. 2021;10(12):5280–94.
Gao L, Zhou F. Comprehensive analysis of RUNX and TGF-β mediated regulation of immune cell infiltration in breast cancer. Front Cell Dev Biol. 2021;9: 730380.
Wu D, Yin Z, Ji Y, Li L, Li Y, Meng F, et al. Identification of novel autophagy-related lncRNAs associated with a poor prognosis of colon adenocarcinoma through bioinformatics analysis. Sci Rep. 2021;11(1):8069.
Yu L, Shen H, Ren X, Wang A, Zhu S, Zheng Y, et al. Multi-omics analysis reveals the interaction between the complement system and the coagulation cascade in the development of endometriosis. Sci Rep. 2021;11(1):11926.
Liu Y, Wang J, Li L, Qin H, Wei Y, Zhang X, et al. AC010973.2 promotes cell proliferation and is one of six stemness-related genes that predict overall survival of renal clear cell carcinoma. Sci Rep. 2022;12(1):4272.
Wei X, Dong Y, Chen X, Ren X, Li G, Wang Y, et al. Construction of circRNA-based ceRNA network to reveal the role of circRNAs in the progression and prognosis of metastatic clear cell renal cell carcinoma. Aging. 2020;12(23):24184–207.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400-16.e11.
Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22.
Martens JW, Margossian AL, Schmitt M, Foekens J, Harbeck N. DNA methylation as a biomarker in breast cancer. Future Oncol. 2009;5(8):1245–56.
Modhukur V, Iljasenko T, Metsalu T, Lokk K, Laisk-Podar T, Vilo J. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 2018;10(3):277–88.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437-1452.e17.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Miao YR, Zhang Q, Lei Q, Luo M, Xie GY, Wang H, et al. ImmuCellAI: a unique method for comprehensive T-cell subsets abundance prediction and its application in cancer immunotherapy. Adv Sci. 2020;7(7):1902880.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER20 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–14.
Pouliot MC, Labrie Y, Diorio C, Durocher F. The role of methylation in breast cancer susceptibility and treatment. Anticancer Res. 2015;35(9):4569–74.
Basu A, Ramamoorthi G, Jia Y, Faughn J, Wiener D, Awshah S, et al. Immunotherapy in breast cancer: current status and future directions. Adv Cancer Res. 2019;143:295–349.
McDonald KA, Kawaguchi T, Qi Q, Peng X, Asaoka M, Young J, et al. Tumor heterogeneity correlates with less immune response and worse survival in breast cancer patients. Ann Surg Oncol. 2019;26(7):2191–9.
Januškevičienė I, Petrikaitė V. Heterogeneity of breast cancer: the importance of interaction between different tumor cell populations. Life Sci. 2019;239:117009.
Khazaeli Najafabadi M, Mirzaeian E, Memar Montazerin S, Tavangar AR, Tabary M, Tavangar SM. Role of GATA3 in tumor diagnosis: a review. Pathol Res Pract. 2021;226:153611.
Heino S, Fang S, Lähde M, Högström J, Nassiri S, Campbell A, et al. Lef1 restricts ectopic crypt formation and tumor cell growth in intestinal adenomas. Sci Adv. 2021;7(47):eabj0512.
Jin Y, Qi G, Chen G, Wang C, Fan X. Association between B-Myb proto-oncogene and the development of malignant tumors. Oncol Lett. 2021;21(2):166.
Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 2019;176(1–2):98-112.e14.
Xu K, Zhang W, Wang C, Hu L, Wang R, Wang C, et al. Integrative analyses of scRNA-seq and scATAC-seq reveal CXCL14 as a key regulator of lymph node metastasis in breast cancer. Hum Mol Genet. 2021;30(5):370–80.
López-Menéndez C, Vázquez-Naharro A, Santos V, Dubus P, Santamaría PG, Martínez-Ramírez Á, et al. E2A modulates stemness, metastasis, and therapeutic resistance of breast cancer. Cancer Res. 2021;81(17):4529–44.
Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32(5):669-83.e5.
Chen H, Ma X, Yang M, Wang M, Li L, Huang T. Transcription factor profiling to predict recurrence-free survival in breast cancer: development and validation of a nomogram to optimize clinical management. Front Genet. 2020;11:333.
Zhou M, Zhong L, Xu W, Sun Y, Zhang Z, Zhao H, et al. Discovery of potential prognostic long non-coding RNA biomarkers for predicting the risk of tumor recurrence of breast cancer patients. Sci Rep. 2016;6:31038.
Bao X, Anastasov N, Wang Y, Rosemann M. A novel epigenetic signature for overall survival prediction in patients with breast cancer. J Transl Med. 2019;17(1):380.
Mao XY, Lee MJ, Zhu J, Zhu C, Law SM, Snijders AM. Genome-wide screen identifies a novel prognostic signature for breast cancer survival. Oncotarget. 2017;8(8):14003–16.
Sun J, Chen X, Wang Z, Guo M, Shi H, Wang X, et al. A potential prognostic long non-coding RNA signature to predict metastasis-free survival of breast cancer patients. Sci Rep. 2015;5:16553.
Wu Y, Liu Z, Wei X, Feng H, Hu B, Liu B, et al. Identification of the functions and prognostic values of RNA binding proteins in bladder cancer. Front Genet. 2021;12: 574196.
Guo C, Shao T, Jiang X, Wei D, Wang Z, Li M, et al. Comprehensive analysis of the functions and prognostic significance of RNA-binding proteins in bladder urothelial carcinoma. Am J Transl Res. 2020;12(11):7160–73.
Fan C, Wang Q, van der Zon G, Ren J, Agaser C, Slieker RC, et al. OVOL1 inhibits breast cancer cell invasion by enhancing the degradation of TGF-β type I receptor. Signal Transduct Target Ther. 2022;7(1):126.
Iqbal MA, Siddiqui S, Ur Rehman A, Siddiqui FA, Singh P, Kumar B, et al. Multiomics integrative analysis reveals antagonistic roles of CBX2 and CBX7 in metabolic reprogramming of breast cancer. Mol Oncol. 2021;15(5):1450–65.
Ma C, Zu X, Liu K, Bode AM, Dong Z, Liu Z, et al. Knockdown of pyruvate kinase m inhibits cell growth and migration by reducing NF-kB activity in triple-negative breast cancer cells. Mol Cells. 2019;42(9):628–36.
Blazquez R, Rietkötter E, Wenske B, Wlochowitz D, Sparrer D, Vollmer E, et al. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int J Cancer. 2020;146(11):3170–83.
Yoda T, McNamara KM, Miki Y, Onodera Y, Takagi K, Nakamura Y, et al. KLF15 in breast cancer: a novel tumor suppressor? Cell Oncol. 2015;38(3):227–35.
Inoue A, Omoto Y, Yamaguchi Y, Kiyama R, Hayashi SI. Transcription factor EGR3 is involved in the estrogen-signaling pathway in breast cancer cells. J Mol Endocrinol. 2004;32(3):649–61.
Peng Y, Xi X, Li J, Ni J, Yang H, Wen C, et al. miR-301b and NR3C2 co-regulate cells malignant properties and have the potential to be independent prognostic factors in breast cancer. J Biochem Mol Toxicol. 2021;35(2):e22650.
Gasparyan M, Lo MC, Jiang H, Lin CC, Sun D. Combined p53- and PTEN-deficiency activates expression of mesenchyme homeobox 1 (MEOX1) required for growth of triple-negative breast cancer. J Biol Chem. 2020;295(34):12188–202.
Lee JS, Oh Y, Park JH, Kyung SY, Kim HS, Yoon S. Terconazole, an azole antifungal drug, increases cytotoxicity in antimitotic drug-treated resistant cancer cells with substrate-specific P-gp inhibitory activity. Int J Mol Sci. 2022;23(22):13809.
Fuseya S, Yamamoto K, Minemura H, Yamaori S, Kawamata T, Kawamata M. Systemic QX-314 reduces bone cancer pain through selective inhibition of transient receptor potential vanilloid subfamily 1-expressing primary afferents in mice. Anesthesiology. 2016;125(1):204–18.
Massari NA, Nicoud MB, Medina VA. Histamine receptors and cancer pharmacology: an update. Br J Pharmacol. 2020;177(3):516–38.
Tower H, Ruppert M, Britt K. The immune microenvironment of breast cancer progression. Cancers. 2019;11(9):1375.
Bense RD, Sotiriou C, Piccart-Gebhart MJ, Haanen JBAG, van Vugt MATM, de Vries EGE, et al. Relevance of tumor-infiltrating immune cell composition and functionality for disease outcome in breast cancer. J Natl Cancer Inst. 2016;109(1):djw192.
Jiang Y, Zhang Q, Hu Y, Li T, Yu J, Zhao L, et al. ImmunoScore signature: a prognostic and predictive tool in gastric cancer. Ann Surg. 2018;267(3):504–13.
Burugu S, Asleh-Aburaya K, Nielsen TO. Immune infiltrates in the breast cancer microenvironment: detection, characterization and clinical implication. Breast Cancer. 2017;24(1):3–15.
Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–47.
Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234(6):8509–21.
Wang M, Wang S, Desai J, Trapani JA, Neeson PJ. Therapeutic strategies to remodel immunologically cold tumors. Clin Transl Immunol. 2020;9(12): e1226.
Zhang J, Huang D, Saw PE, Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol. 2022;43(7):523–45.
Prendergast GC, Mondal A, Dey S, Laury-Kleintop LD, Muller AJ. Inflammatory reprogramming with IDO1 inhibitors: turning immunologically unresponsive “Cold” tumors “Hot.” Trends Cancer. 2018;4(1):38–58.
Funding
This work was supported by Youth Scientific Research Project of Fujian Provincial Health Commission (No. 2020QNA046).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and data collection and analysis were performed by QW, SZ, NL and XX. The first draft of manuscript was written by QW and XX with all authors commenting on its previous versions, after which all the above mentioned authors read and approved the final manuscript.
Registration and protocol
The review was not registered with protocol not yet prepared.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
R code and data.
Additional file 2. Table S1.
The TF family genes.
Additional file 3: Figure S1.
Kaplan–Meier curves for OS prediction in TCGA-BRCA of A MEOX1; B RGCC; C ZNF106; D BHLHE41; E LEF1; F NFKBIE; G NR3C2; H KLF15; I OVOL1; J CBX2; K EGR3.
Additional file 4: Figure S2.
Kaplan–Meier curves for OS prediction in BRCA subtypes of A Her2 or Basal and B M1.
Additional file 5: Figure S3.
Heatmap of DNA methylation expression levels of the TFDEGs in BRCA by MethSurv platform.
Additional file 6: Table S2.
Prognostic Value of Single CpG of the TFDEGs in BRCA by MethSurv platform.
Additional file 7: Figure S4.
Correlations between expressions of TFDEGs and immune infiltration profiles in BRCA. The figure shows the expression of each gene associated with tumor purity and several tumor-infiltrating immune cell markers, such as B cell, CD8+ T cell, CD4 + T cell, macrophage, neutrophil, and dendritic cell markers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, Q., Zheng, S., Lin, N. et al. Comprehensive research into prognostic and immune signatures of transcription factor family in breast cancer. BMC Med Genomics 16, 87 (2023). https://doi.org/10.1186/s12920-023-01521-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01521-y