
Abstract
Background
Noonan syndrome (NS) is a genetic disorder characterized by developmental delays, typical facial gestalt and cardiovascular defects. LZTR1 variants have been recently described in patients with NS and schwannomatosis, but the association, inheritance pattern and management strategy has not been fully elucidated. Here, we review the contribution of LZTR1 in NS and describe a patient with a novel, likely pathogenic variant in LZTR1.
Case presentation
A female patient was diagnosed with clinical NS at 8 months of age. She presented in adulthood when a brain and spine MRI identified plexiform neurofibromas; however, she did not meet the clinical criteria for Neurofibromatosis type 1. No pathogenic variants were identified through molecular genetic analysis of NF1, SPRED1 and a multigene NS panel. Whole exome sequencing at age 23 identified a novel de novo likely pathogenic heterozygous variant in the LZTR1 gene denoted as c.743G>A (p.Gly248Glu). Serial MRIs have shown stable imaging findings and the patient is being followed clinically by cardiology, neurology and medical genetics.
Conclusions
We identified a novel mutation in the LZTR1 gene, not previously reported in association with NS. This report provides additional evidence to support for the assessment of schwannomatosis in patients with LZTR1-NS and may have overlap with Neurofibromatosis type 1.
Similar content being viewed by others


Background
Noonan syndrome (NS) is a genetic multisystem disorder with a prevalence of 1 in 1000–2500 live births (1). This condition is characterised by varying developmental delays, distinctive facial features, congenital heart defects and short stature; clinical diagnosis is often based on these features (2). NS is part of a group of phenotypically similar developmental disorders (RASopathies), caused by germline variants in the genes within the RAS/MAPK signalling pathway (3).
There are multiple genes that cause NS, all linked to the RAS/MAPK signalling pathway (4, 5). Fifty percent of individuals with NS have a germline pathogenic variant (PV) in PTPN11 (2). Other reported genes include SOS1, RAF1, ROT1 and KRAS, occurring in 13%, 5%, 5% and < 5% of cases, respectively (2). PVs in other genes, including BRAF, MAP2K1 and NRAS, have been identified in less than 1% of affected individuals (2). Another gene, LZTR1, has been recently identified as a causative gene in RASopathies (6); however, its precise role in the RAS/MAPK signalling pathway is less defined. Additionally, germline pathogenic variants in LZTR1 have been identified in patients with schwannomatosis and is thought to be a distinct entity as schwannomas are not frequently seen in NS (7, 8). There is a paucity of cases to address this genetic and phenotypic heterogeneity in LZTR1 carriers.
LZTR1 (OMIM 600574), encoding leucine zipper-like transcription regulator 1 (LZTR1), was proposed as a tumor suppressor gene belonging to the BTB-Kelch superfamily (9). LZTR1 is a Golgi protein and belongs to the BTB-Kelch superfamily (7, 9), where it is reported to be involved in apoptosis (9) and acts as a substrate-specific adaptor for the Cullin-3 (Cul3) ubiquitin ligase (10, 11). Using a computational platform, LZTR1 was identified as a tumor suppressor gene, with somatic mutations in this gene driving glioblastoma (10). Somatic mutations in LZTR1 have also been associated with liver cancer (12).
Recent investigations have provided more insight into its potential role in the RAS/MAPK signalling pathway. LZTR1 binds to the RAF1/SHOC2/PP1CB complex and promotes RAF1 Ser259 phosphorylation, leading to MAPK signalling pathway inactivation (13). Additional data reports that LZTR1 enables the polyubiquitination and degradation of endogenous RAS, which ultimately inhibits RAS/MAPK signalling (14). Two other studies suggest that LZTR1 mediates RAS ubiquitination and MAPK pathway activation, contributing to the development of human disease (15, 16). Examination of LZTR1 variants associated with NS suggest this gene is functionally-linked to the RAS/MAPK pathway by negatively controlling RAS protein levels and MAPK signalling (17). In addition, a biological relationship has been proposed between LZTR1 and RIT1, whereby pathogenic mutations affecting RIT1 or LZTR1 leads to RIT1 accumulation and contributes to hyperactivation of MAPK signalling (18).
While the association of germline LZTR1 variants with human disease is still being elucidated, germline loss-of-function mutations in LZTR1 predispose to schwannomatosis (7, 19, 20) and NS (2). NS has wide genetic heterogeneity and clinical variability (21). Inheritance most frequently occurs in an autosomal dominant (AD) manner, however, PVs in LZTR1 leading to NS can also be inherited in an autosomal recessive (AR) manner (2). Expression experiments suggest that LZTR1 variants that cause AD NS may not be gain-of-function, whereas variants in patients with AR NS may have a loss-of-function effect (13). Loss-of-function mutations in biallelic LZTR1 variants include splice site, frameshift, nonsense and missense modifications (17). Experiments assessing the functionality of missense LZTR1 mutations suggests that schwannomatosis-associated LZTR1 mutations act heterogeneously in modifying RAS-MAPK signalling, similar to variants causing dominant NS (17). The pleotropic function of LZTR1 may explain the multiple inheritance patterns (AR, AD) of LZTR1-associated NS. NS has been associated with Neurofibromatosis type 1 (NF1) [Neurofibromatosis-Noonan syndrome (NFNS)], a rare disorder where individuals present with phenotypic characteristics of these two autosomal dominant conditions (22). While there was an earlier debate about whether NFNS is a separate genetic condition, more recent reports illustrated clinical findings of both disorders, providing support that this is a new syndrome (23). NFNS is mainly caused due to mutations in NF1 (24–28), however, there has occasionally been a PTPN11 mutation reported in addition to the NF1 gene mutation (29, 30). The involvement of other genes within the RAS-MAPK signalling pathway that cause NS have not yet been explored in NFNS (31). As such, NS should be considered part of the differential diagnosis in NF1 patients.
In this report, we describe a patient with clinically diagnosed NS who presented with sacral plexiform nerve sheath tumors, suggestive of neurofibromas or schwannomas. Whole exome sequencing (WES) revealed a de novo likely pathogenic heterozygous variant in the LZTR1 gene. Here we demonstrate the clinical overlap of NS, schwannomatosis and NF1 and support the inclusion of LZTR1 in gene panels for NS. In addition, we have reviewed the published literature on NS patients and variants in the LZTR1 gene, noting zygosity, inheritance pattern and clinical characteristics, to inform the management of individuals with a heterozygous LZTR1 mutation.
Case presentation
Case description
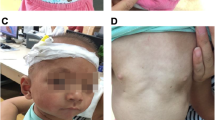
A Caucasian female received a clinical diagnosis of NS at 8 months of age, prompted by an extra nuchal skin fold (Fig. 1). Facial features were consistent with NS (large eyes, low-set cupped ears and prominent lips) and she was also found to have congenital heart defects (subaortic ridge with mild left ventricular outflow tract obstruction, mitral valve prolapse with mild mitral regurgitation and tricuspid aortic valve with mild aortic insufficiency), recurrent migraines, pectus excavatum and a history of learning disability.

Timeline of the patient’s diagnosis, symptoms and treatment
The patient received daily growth hormone (GH) injections as a child between ages 9–16 to address short stature. Her pre-growth hormone height was 118.6 cm (< 5%ile according to CDC growth charts and 50%ile in Noonan Syndrome growth charts) (32). No significant adverse effects were reported. Her height at adulthood (~ 17 years of age) was 158.4 cm, placing her in the 25%ile on the CDC Growth Charts and 90%ile on Noonan Syndrome growth charts.
Due to delayed puberty, the patient had a trial of transdermal estradiol at the age of 14. This was transitioned to Premarin with good gonadotropic effect, and she has been on oral contraceptives since 16 years of age.
At 20 years of age, a magnetic resonance imaging (MRI) brain and intracranial magnetic resonance angiography (MRA) was performed for chronic frontal headaches. MRA was normal. MRI of the brain and subsequent MRIs of the whole spine revealed a cerebellar ectopia related to a Chiari type 1 malformation and septated syringomelia extending from C1-T6 with no evidence of an underlying mass. There were multiple plexiform neurofibromas in the sacral spine, prompting a referral to neurosurgery for suspicion of NF1. At 23 years of age, a discontinuous expansile syrinx present from C6-T6 was noted, with enlargement of the dorsal root ganglia in the cervical spine.
The plexiform neurofibromas involved all nerve roots and caused marked expansion of the sacral foramina. As a biopsy was unlikely to change management, these lesions were not biopsied and thus imaging findings were not pathologically confirmed (Fig. 2A–B). Serial MRIs have shown stable imaging findings. Upon clinical examination, the patient did not present with café-au-lait macules (CALMs), skinfold freckling, or cutaneous or subcutaneous neurofibromas. Due to the initial question of NF1, genetic testing was conducted in a step wise fashion to establish a molecular genetic diagnosis.

De-identified images of the patient’s plexiform neurofibromas. Panel A shows cornal T1 sequence, with multiple hypodense lesions arising from the sacral nerve roota, in keeping with neurofibromas (white arrows). Panel B shows sagittal T2 sequence, with neurofibromas seen as hyperintense lesions (white arrows)
Next generation sequencing
Exon-level germline analysis of NF1 [NCBI RefSeq NM_000267.3], SPRED1 [NM_152594.2] and a multigene NS panel (BRAF [NM_004333.4], CBL [NM_005188.3], HRAS [NM_005343.2], KRAS [NM_004985.3], MAP2K1 [NM_002755.3], MAP2K2 [NM_030662.3], NRAS [NM_002524.4], PTPN11 [NM_002834.3], RAF1 [NM_002880.3], RIT1 [NM_006912.5], SHOC2 [NM_007373.3], SOS1 [NM_005633.3]) was completed on DNA extracted from blood leukocytes at The Hospital for Sick Children Genome Diagnostics Laboratory (Toronto, ON). Next Generation Sequencing (NGS) was performed using a targeted Agilent SureSelect custom capture followed by paired-end sequencing using the Illumina sequencing platform. Variant calls were generated using Genomic Analysis Tool Kit (GATK) after read alignment with the Burrows-Wheeler Aligner (BWA). Genome build NCBI37/hg19 with decoy and data analysis software SK High Coverage Clinical Pipeline was used. Multiplex ligation-dependent probe amplification (MLPA) was used to test gene dosage.
Clinical exome sequencing analysis (NGS with copy number variant [CNV]) was completed by the GeneDx Molecular Laboratory (Gaithersburg, USA) on DNA obtained via a buccal swab. The enriched targets were simultaneously sequenced with paired-end reads on an Illumina platform and genome build GRCh37/UCSC hg19. Using a custom-developed analysis tool (XomeAnalyzer), data were filtered and analysed to identify sequence variants and most deletions and duplications involving three or more coding exons (33). Sequence and CNVs are reported according to the Human Genome Variation Society (HGVS) or International System for Human Cytogenetic Nomenclature (ISCN) guidelines, respectively. Reportable PVs, likely PVs and variants of uncertain significance were reported as per AMP/ACMG guidelines (34). Secondary findings were examined in the coding regions of the gene list provided by ACMG SF v2.0 (September 2016) (35).
Molecular genetic approach
Exon-level germline genetic testing of the NF1, SPRED1, and a multigene NS panel that did not include the LZTR1 gene did not reveal any PVs in the genes tested. A single nucleotide polymorphism (SNP) microarray at the same laboratory was in-keeping with a normal female, arr(1-22,X)×2. As schwannomas and neurofibromas are not typically reported in patients with NS, the molecular genetic approach in patients with nerve sheath tumors often begins with NF1. For this patient, multi-gene panel testing for NS, NF1 and Legius syndrome were negative and a negative SNP chromosomal microarray. Of note, due to the emerging literature for LZTR1 in NS, LZTR1 was not yet included in the laboratory’s NS gene panel.
Clinical exome sequencing analysis identified a likely pathogenic heterozygous c.743G>A (p.Gly248Glu) in exon 8 of the LZTR1 gene [NM_006767.3]. Variants at this residue are normally associated with an autosomal dominant disorder. These findings are consistent with the patient’s reported clinical features. No reportable secondary findings were identified and was in accordance to the reporting structure recommended by the American College of Medical Genetics (ACMG) (35, 36).
The p.Gly248Glu variant identified in this study is located in the Kelch domain of LZTR1; heterozygous missense mutations in this region have been seen in patients with a clinical diagnosis of NS (6), NS patients with heterozygous PVs in LZTR1 (37), as well as in NS patients with a bleeding phenotype (8, 38). This variant has previously been identified in a patient with fetal pleural effusion and NS (39) and has not been observed in larger population cohorts (40).
Parental targeted variant testing confirmed this was a de novo variant in the patient. Her parents have no features of NS, supporting the causality of this variant. The patient and her family have undergone genetic counselling regarding implications of the results for her, her family and any future children. She is being followed clinically by cardiology, neurology and medical genetics.
Discussion and conclusions
Monoallelic and biallelic LZTR1 variants in NS are not well described, however, previous studies have reported germline variants causative for NS (Table 1) in either an autosomal recessive or autosomal dominant manner (6, 8, 13, 37, 38, 41–47). Of note, a different missense mutation at the same protein residue as our patient (p.Gly248Arg) was reported as pathogenic or likely pathogenic in other individuals with NS (6, 13, 38, 47), with the same designations on ClinVar. In silico analysis of variants (p.Ala116Val, p.Arg284Cys, p.Arg688Cys, p.Gly248Arg, p.His287Tyr, p.Pro520Leu, p.Ser247Asn, p.Ser122Leu, p.Tyr119Cys and p.Val456Gly) found in the LZTR1 gene support a deleterious effect (6, 7, 42).
Our patient’s phenotype is unique, as not many patients with NS present with plexiform nerve sheath tumors. These are typically benign tumors, with neurofibromas characteristic of NF1 (48) and schwannomas often occurring in patients with NF2 or schwannomatosis (49). This case highlights one of the main issues in identifying NS patients—there is a degree of phenotypic overlap and uncertainty between other similar conditions; therefore, the focus of genetic testing may be on this rare manifestation with a misdiagnosis of NF1, NF2 or schwannomatosis, rather than NS. A recent study performed an extensive literature review to estimate the number of conditions that may mimic NF1, as well as examining data from 40 pediatric patients with NF1-like syndromes (50). Phenotypic overlap, particularly with skin manifestations or tissue overgrowth, was observed between NF1, NF1-like syndromes, the RASopathies and other disorders associated with higher tumor development, including phosphatase and tensin homolog (PTEN) hamartoma tumor syndromes, constitutional mismatch repair deficiency (CMMRD) syndromes, chromosomal abnormalities and multiple endocrine neoplasia (MEN) syndromes (50). Furthermore, genotypic overlap can also be seen between NF1-like syndrome and other disorders, including NS (50).
Besides our patient, there have been a few reports of neurofibromas/schwannomas in patients with a RASopathy phenotype. One patient presented with an overgrowth of peripheral nerve sheaths, suggestive of a schwannoma or neurofibroma (51). The final diagnosis was multiple diffuse schwannomas; although this is suggestive of NF2 or schwannomatosis, a germline KRAS variant (p.Lys5Glu) was identified (51). In one family with unaffected parents and four affected children with NS, a c.2220-17C>A (p.Tyr741Hisfs*89) maternally-inherited splice variant was found; several members of this family had suggestive signs of schwannomas on MRI analysis (37). Another individual with a c.740C>A (p.Ser247Asn) variant in LZTR1 developed multiple schwannomas in the right arm, however, no material from the schwannomas was available for molecular testing (6). This individual was the mother of a patient with NS (6).
Carriers of LZTR1 have phenotypic heterogeneity and management of LZTR1-NS patients has not been well described. Guidelines regarding the management of individuals with NS varies and is based on the clinical manifestations. Some common treatments include: treatment for cardiovascular anomalies, early intervention programs for developmental disabilities, treatment for serious bleeding conditions, GH treatment for short stature and further monitoring for abnormalities (2). In comparison, germline or mosaic mutations in the LZTR1 (7, 19, 20) and the SMARCB1 (52) genes have been associated with schwannomatosis, although the link between LZTR1 schwannomatosis and the development of other tumors has not been clearly defined. Current clinical management for schwannomatosis recommends that individuals have a baseline brain and spine MRI in late childhood/early adulthood to monitor the disease and management in adulthood should be performed by a neurologist or neurofibromatosis specialist to manage pain (53). Whole body MRI and increased surveillance can be considered if the patient is symptomatic (53). The guidelines on how to manage LZTR1-NS and LZTR1-schwannamatosis patients are not clear; therefore, a reasonable approach would be management of patients under both NS and schwannomatosis guidelines. Clarification of the genetic etiology of NS should be pursued early in the course of NS management as this may have implications on the use of GH therapy in a patient with LZTR1-NS. This includes consideration of tumor development and cardiac abnormalities, as individuals with NS are at increased risk, and there have been reports of tumors (54–57) and adverse cardiac reactions (58) following GH therapy.
Our proband’s likely PV in the LZTR1 gene was not detected on the original NF1, SPRED1 and multigene NS panel. Eventually, clinical exome sequencing analysis was able to identify the causative gene, which was later confirmed to be absent in her clinically unaffected parents. In 2018, the Clinical Genome Resource (ClinGen) RASopathy expert panel assessed 19 genes, including LZTR1, and found a strong association between LZTR1 and AD NS (59). As of 2020, the RASopathy expert panel definitively associates LZTR1 with AD NS and has stated there is strong evidence between LZTR1 and AR NS. Since then, LZTR1 has been a common new addition to many commercial and academic and laboratory NS panels. Based on our findings, inclusion of additional NS genes to schwannomatosis panels could also be considered. Due to the high de novo rate of variants causing NF1, NS and schwannomatosis, an alterative approach may be trio whole exome sequencing to aid in the interpretation of variants.
Based on previous reports and our findings, individuals with NS and a monoallelic or biallelic germline LZTR1 mutation may be at an increased risk of developing schwannomas and may meet the diagnostic criteria for schwannomatosis. Additional case reports of NS and variants in LZTR1, as well as functional studies showing the involvement of LZTR1 in the RAS/MAPK pathway would provide support for the creation of management guidelines for LZTR1-NS individuals. Due to the difficulties with overlapping phenotypes and genotypes, we believe that all NF1-like syndromes should be assessed in young individuals when identifying a causative gene and disorder. As clinical guidelines and gene panels change, clinicians should perform annual assessments of these patients to re-evaluate their original diagnosis and management. Due to the clinical implications for this patient presenting with schwannomas and NS, our patient will be followed as per NS and schwannomatosis surveillance guidelines to screen for nervous system tumors and monitor for tumor development.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study. The sequencing data that support the findings of this study will be made available on ClinVar under the following accession number: SCV002106410.
Abbreviations
- NS:
-
Noonan syndrome
- PV:
-
Pathogenic variant
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- MRI:
-
Magnetic resonance imaging
- MRA:
-
Magnetic resonance angiography
- NF1:
-
Neurofibromatosis type 1
- NF2:
-
Neurofibromatosis type 2
- CALMs:
-
Café-au-lait macules
- SNP:
-
Single nucleotide polymorphism
- CNV:
-
Copy number variant
- NGS:
-
Next generation sequencing
References
Mendez HM, Opitz JM. Noonan syndrome: a review. Am J Med Genet. 1985;21(3):493–506.
Allanson JE, Roberts AM. Noonan syndrome. Available from https://www.ncbi.nlm.nih.gov/books/NBK1124/: GeneReviews; 2019.
Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19(3):230–6.
Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308.
Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci. 2009;100(10):1786–93.
Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52(6):413–21.
Piotrowski A, Xie J, Liu YF, Poplawski AB, Gomes AR, Madanecki P, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. 2014;46(2):182–7.
Jacquinet A, Bonnard A, Capri Y, Martin D, Sadzot B, Bianchi E, et al. Oligo-astrocytoma in LZTR1-related Noonan syndrome. Eur J Med Genet. 2020;63(1): 103617.
Nacak TG, Leptien K, Fellner D, Augustin HG, Kroll J. The BTB-kelch protein LZTR-1 is a novel Golgi protein that is degraded upon induction of apoptosis. J Biol Chem. 2006;281(8):5065–71.
Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–9.
Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Privé GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6(10):R82.
Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327-41.e23.
Umeki I, Niihori T, Abe T, Kanno SI, Okamoto N, Mizuno S, et al. Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1-PPP1CB complexes. Hum Genet. 2019;138(1):21–35.
Abe T, Umeki I, Kanno SI, Inoue SI, Niihori T, Aoki Y. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 2020;27(3):1023–35.
Bigenzahn JW, Collu GM, Kartnig F, Pieraks M, Vladimer GI, Heinz LX, et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science. 2018;362(6419):1171–7.
Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science. 2018;362(6419):1177–82.
Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet. 2019;28(6):1007–22.
Castel P, Cheng A, Cuevas-Navarro A, Everman DB, Papageorge AG, Simanshu DK, et al. RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science. 2019;363(6432):1226–30.
Paganini I, Chang VY, Capone GL, Vitte J, Benelli M, Barbetti L, et al. Expanding the mutational spectrum of LZTR1 in schwannomatosis. Eur J Hum Genet. 2015;23(7):963–8.
Smith MJ, Isidor B, Beetz C, Williams SG, Bhaskar SS, Richer W, et al. Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology. 2015;84(2):141–7.
Allanson JE, Hall JG, Hughes HE, Preus M, Witt RD. Noonan syndrome: the changing phenotype. Am J Med Genet. 1985;21(3):507–14.
Opitz JM, Weaver DD. The neurofibromatosis-Noonan syndrome. Am J Med Genet. 1985;21(3):477–90.
Işık E, Onay H, Atik T, Solmaz AE, Özen S, Çoğulu Ö, et al. A Neurofibromatosis Noonan syndrome patient presenting with abnormal external genitalia. J Clin Res Pediatr Endocrinol. 2020;12(1):113–6.
De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet. 2005;77(6):1092–101.
Baralle D, Mattocks C, Kalidas K, Elmslie F, Whittaker J, Lees M, et al. Different mutations in the NF1 gene are associated with Neurofibromatosis-Noonan syndrome (NFNS). Am J Med Genet A. 2003;119a(1):1–8.
Hüffmeier U, Zenker M, Hoyer J, Fahsold R, Rauch A. A variable combination of features of Noonan syndrome and neurofibromatosis type I are caused by mutations in the NF1 gene. Am J Med Genet A. 2006;140(24):2749–56.
Zhang Z, Chen X, Zhou R, Yin H, Xu J. Chinese patient with neurofibromatosis-Noonan syndrome caused by novel heterozygous NF1 exons 1–58 deletion: a case report. BMC Pediatr. 2020;20(1):190.
Stevenson DA, Viskochil DH, Rope AF, Carey JC. Clinical and molecular aspects of an informative family with neurofibromatosis type 1 and Noonan phenotype. Clin Genet. 2006;69(3):246–53.
Bertola DR, Pereira AC, Passetti F, de Oliveira PS, Messiaen L, Gelb BD, et al. Neurofibromatosis-Noonan syndrome: molecular evidence of the concurrence of both disorders in a patient. Am J Med Genet A. 2005;136(3):242–5.
D'Amico A, Rosano C, Pannone L, Pinna V, Assunto A, Motta M, et al. Clinical variability of neurofibromatosis 1: a modifying role of cooccurring PTPN11 variants and atypical brain MRI findings. Clin Genet. 2021.
Nyström AM, Ekvall S, Allanson J, Edeby C, Elinder M, Holmström G, et al. Noonan syndrome and neurofibromatosis type I in a family with a novel mutation in NF1. Clin Genet. 2009;76(6):524–34.
Malaquias AC, Brasil AS, Pereira AC, Arnhold IJ, Mendonca BB, Bertola DR, et al. Growth standards of patients with Noonan and Noonan-like syndromes with mutations in the RAS/MAPK pathway. Am J Med Genet A. 2012;158a(11):2700–6.
Retterer K, Scuffins J, Schmidt D, Lewis R, Pineda-Alvarez D, Stafford A, et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med. 2015;17(8):623–9.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–74.
Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med. 2018;20(10):1175–85.
Pagnamenta AT, Kaisaki PJ, Bennett F, Burkitt-Wright E, Martin HC, Ferla MP, et al. Delineation of dominant and recessive forms of LZTR1-associated Noonan syndrome. Clin Genet. 2019;95(6):693–703.
Weissbach T, Kushnir A, Rasslan R, Rosenblatt O, Yinon Y, Berkenstadt M, et al. Fetal pleural effusion: contemporary methods of genetic evaluation. Prenat Diagn. 2019;39(9):751–7.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Ferrari L, Mangano E, Bonati MT, Monterosso I, Capitanio D, Chiappori F, et al. Digenic inheritance of subclinical variants in Noonan Syndrome patients: an alternative pathogenic model? Eur J Hum Genet. 2020;28(10):1432–45.
Ghedira N, Kraoua L, Lagarde A, Abdelaziz RB, Olschwang S, Desvignes JP, et al. Further evidence for the implication of LZTR1, a gene not associated with the Ras-Mapk pathway, in the pathogenesis of Noonan syndrome. Biol Med. 2017;9(6).
Chen H, Li X, Liu X, Wang J, Zhang Z, Wu J, et al. Clinical and mutation profile of pediatric patients with RASopathy-associated hypertrophic cardiomyopathy: results from a Chinese cohort. Orphanet J Rare Dis. 2019;14(1):29.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC Endocr Disord. 2021;21(1):2.
Perin F, Trujillo-Quintero JP, Jimenez-Jaimez J, Rodríguez-Vázquez Del Rey MDM, Monserrat L, Tercedor L. Two novel cases of autosomal recessive Noonan syndrome associated with LZTR1 variants. Rev Esp Cardiol (Engl Ed). 2019;72(11):978–80.
Nakaguma M, Jorge AAL, Arnhold IJP. Noonan syndrome associated with growth hormone deficiency with biallelic LZTR1 variants. Genet Med. 2019;21(1):260.
Güemes M, Martín-Rivada Á, Ortiz-Cabrera NV, Martos-Moreno G, Pozo-Román J, Argente J. LZTR1: genotype expansion in Noonan syndrome. Horm Res Paediatr. 2019;92(4):269–75.
Ferner RE, O’Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15(6):679–84.
Westhout FD, Mathews M, Paré LS, Armstrong WB, Tully P, Linskey ME. Recognizing schwannomatosis and distinguishing it from neurofibromatosis type 1 or 2. J Spinal Disord Tech. 2007;20(4):329–32.
Pinti E, Nemeth K, Staub K, Lengyel A, Fekete G, Haltrich I. Diagnostic difficulties and possibilities of NF1-like syndromes in childhood. BMC Pediatr. 2021;21(1):331.
Bertola DR, Pereira AC, Brasil AC, Suzuki L, Leite C, Falzoni R, et al. Multiple, diffuse schwannomas in a RASopathy phenotype patient with germline KRAS mutation: a causal relationship? Clin Genet. 2012;81(6):595–7.
Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80(4):805–10.
Evans DGR, Salvador H, Chang VY, Erez A, Voss SD, Druker H, et al. Cancer and central nervous system tumor surveillance in pediatric Neurofibromatosis 2 and related disorders. Clin Cancer Res. 2017;23(12):e54–61.
McWilliams GD, SantaCruz K, Hart B, Clericuzio C. Occurrence of DNET and other brain tumors in Noonan syndrome warrants caution with growth hormone therapy. Am J Med Genet A. 2016;170a(1):195–201.
Romano AA, Dana K, Bakker B, Davis DA, Hunold JJ, Jacobs J, et al. Growth response, near-adult height, and patterns of growth and puberty in patients with Noonan syndrome treated with growth hormone. J Clin Endocrinol Metab. 2009;94(7):2338–44.
Osio D, Dahlgren J, Wikland KA, Westphal O. Improved final height with long-term growth hormone treatment in Noonan syndrome. Acta Paediatr. 2005;94(9):1232–7.
Moos D, Droitcourt C, Rancherevince D, Marec Berard P, Skowron F. Atypical granular cell tumor occurring in an individual with Noonan syndrome treated with growth hormone. Pediatr Dermatol. 2012;29(5):665–6.
Wolf CM, Zenker M, Burkitt-Wright E, Edouard T, García-Miñaúr S, Lebl J, et al. Management of cardiac aspects in children with Noonan syndrome - results from a European clinical practice survey among paediatric cardiologists. Eur J Med Genet. 2022;65(1): 104372.
Grant AR, Cushman BJ, Cavé H, Dillon MW, Gelb BD, Gripp KW, et al. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat. 2018;39(11):1485–93.
Acknowledgements
We would like to thank the patient and her family for their participation and support of this study.
Funding
RHK is supported by the Bhalwani Family Charitable Foundation.
Author information
Authors and Affiliations
Contributions
KMF was responsible for drafting the manuscript. ET was the genetic counsellor, CBT and HS were the neurologists and RHK was the medical geneticist providing clinical care for the patient. Plexiform imaging was provided by CBT. ET, CBT, HS and RHK contributed to the interpretation of the findings and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
This study is in approval with the University Health Network Ethics Committee. Written informed consent was obtained from the patient.
Consent for publication
Written informed consent for publication of a case report was obtained from the patient.
Competing interests
CBT has been a consultant for Alexion, CSL, Takeda, Sanofi and has received research grants from Grifols and Octapharma, not related to this work. All other authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Farncombe, K.M., Thain, E., Barnett-Tapia, C. et al. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC Med Genomics 15, 160 (2022). https://doi.org/10.1186/s12920-022-01304-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01304-x