
Abstract
Background
Neurofibromatosis type 1 (NF1), which is caused by heterozygous inactivating pathogenic variants in the NF1, has poor phenotypic expressivity in the early years of life and there are numerous conditions, including many other tumor predisposition syndromes, that can mimic its appearance. These are collectively termed NF1-like syndromes and are also connected by their genetic background. Therefore, the NF1’s clinical diagnostic efficiency in childhood could be difficult and commonly should be completed with genetic testing.
Methods
To estimate the number of syndromes/conditions that could mimic NF1, we compiled them through an extensive search of the scientific literature. To test the utility of NF1’s National Institutes of Health (NIH) clinical diagnostic criteria, which have been in use for a long time, we analyzed the data of a 40-member pediatric cohort with symptoms of the NF1-like syndromes’ overlapping phenotype and performed NF1 genetic test, and established the average age when diagnostic suspicion arises. To facilitate timely identification, we compiled strongly suggestive phenotypic features and anamnestic data.
Results
In our cohort the utility of NF1’s clinical diagnostic criteria were very limited (sensitivity: 80%, specificity: 30%). Only 53% of children with clinically diagnosed NF1 had a detectable NF1 pathogenic variation, whereas 40% of patients without fulfilled clinical criteria tested positive. The average age at first genetic counseling was 9 years, and 40% of children were referred after at least one tumor had already been diagnosed. These results highlight the need to improve NF1-like syndromes’ diagnostic efficiency in childhood. We collected the most extensive spectrum of NF1-like syndromes to help the physicians in differential diagnosis. We recommend the detailed, non-invasive clinical evaluation of patients before referring them to a clinical geneticist.
Conclusions
Early diagnosis of NF1-like syndromes can help to prevent severe complications by appropriate monitoring and management. We propose a potential screening, diagnostic and management strategy based on our findings and recent scientific knowledge.
Similar content being viewed by others


Background
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen’s disease, is caused by heterozygous inactivating pathogenic variations in the NF1 gene. NF1 is the most frequent (prevalence 1–5/10000) neurocutaneous genetic disorder, which is characterized by an autosomal dominant (AD) inheritance pattern with complete penetrance, but very heterogeneous and age-related expressivity [1,2,3]. Its most distinctive features affect the skin, skeletal and nervous system. NF1 is associated with an increased risk for benign and malignant tumors and can be identified by genetic testing or clinical diagnostic criteria. The latter were assembled by The NIH consensus development conference in 1987 and are still in use [4]. The recommendation says that the diagnosis of NF1 should be established if the patient fulfills at least two criteria, or suspected if an individual meets only one of them:
-
≥6 café-au-lait macules (CALMs) (> 5 mm in greatest diameter in prepubertal individuals and > 15 mm in greatest diameter in postpubertal individuals),
-
≥2 neurofibromas of any type or ≥ 1 plexiform neurofibroma,
-
freckling in the axillary or inguinal regions,
-
optic pathway glioma (OPG),
-
≥2 Lisch nodules (iris hamartomas),
-
a distinctive osseous lesion (sphenoid dysplasia or tibial pseudarthrosis),
-
a first-degree relative (parent, sibling, offspring) with NF1 as defined by the above criteria.
The NIH criteria are both highly specific and highly sensitive in adults [5], but not in children. While almost 100% of patients meet the criteria by the age of 20 years, their sensitivity is 97% among eight-year-old children and only 30% by the age of 1 year in infants [6]. Furthermore, there are many other conditions which can mimic the NF1’s appearance and fulfill the abovementioned criteria. Among these NF1-like syndromes there are harmless conditions as well as severe disorders with potential life-threatening complications. They have different inheritance patterns and tumor predisposition effects, so they require diverse screening and therapeutic procedures. Therefore, it is important to identify and distinguish between them in time.
Several previous studies have dealt with the diagnostic challenge of NF1-like conditions [7,8,9,10,11]. Most of them established harmonious recommendations, but the number of open questions remains significant. Some highlighted the significance of certain clinical diagnostic aspects, while others proposed a molecular screening approach. Considering these facts, we attempted to collect all possible NF1-like conditions, evaluated their importance, surveyed the utility of NIH diagnostic criteria on a 40-member-large Hungarian cohort with NF1-like condition under 18 years of age, and analyzed their symptoms. To improve the deficiencies in the field of NF1-like syndromes’ in childhood we propose a possible method of examination, diagnosis, monitoring and management.
Patients and methods
To estimate the number of syndromes/conditions that could mimic NF1, we compiled them through an extensive search of the scientific literature. To test the utility of NF1’s NIH clinical diagnostic criteria, which have been in use for a long time, we analyzed the data of a 40-member pediatric cohort with symptoms of the NF1-like syndromes’ overlapping phenotype and performed NF1 genetic test.
Search for NF1-like syndromes in online databases
With an extended search using the electronic databases PubMed, Online Mendelian Inheritance in Man (OMIM), Google Scholar, Microsoft Academic, Semantic Scholar and the Cochrane Library we attempted to collect all published NF1-like conditions. Every sign, symptom and term possibly associated to NF1 constituted the basis of our search.
Recruitment and clinical characterization of pediatric patients in relation to NF1-like syndromes
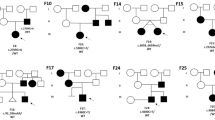
A total of 40 patients (P1–40) under 18 years of age including 24 males (60%) and 16 females (40%) were recruited for this study in relation to the clinically suspected NF1-like syndrome from Semmelweis University’s IInd Department of Pediatrics in Hungary. All children are attended by a licensed clinical geneticist and pediatrician, and undergo exhaustive physical examinations regularly. Age, family tree and patient history were registered during the first counseling and further tests (e.g. brain magnetic resonance imaging (MRI), cardiac ultrasound, endocrinological laboratory tests, etc.) were carried out when necessary. Regular extended medical check-ups include the following specialties/tests: ophthalmology (especially for the presence of Lisch nodules), otolaryngology (objective hearing test), dermatology (appearance and progression of skin lesions), neurology (status and systematic follow-up), abdominal ultrasound (intraabdominal tumors, renal artery stenosis, structure of great abdominal vessels and organs), cardiology (blood pressure measurements, echocardiography), laboratory tests (complete blood count, renal and hepatic function parameters, blood lipids and glucose, inflammatory parameters, iron status, 25-hydroxyvitamin D, calcium and phosphate level, microalbuminuria).
Genetic tests
Every patient underwent exhaustive NF1 variation testing: Giemsa-banding, fluorescent in situ hybridization (FISH), targeted multiplex ligation dependent probe amplification (MLPA), next-generation sequencing (NGS) and Sanger sequencing (Supplementary material 1).
Patients with signs suggestive of a condition other than NF1 were also tested for further gene variations (Supplementary material 2). Analyses of those genes were performed with the combination of FISH, targeted NGS and MLPA involving the entire coding region and flanking splice sites.
The de novo chromosome 4 inversion (46,XX,inv(4)(p13q13)) of a female patient was confirmed with FISH, but the breakpoints were not refined, because her family declined further genetic testing.
In every case all genetic tests were carried out on genomic deoxyribonucleic acid (DNA) isolated from peripheral blood sample.
Patients with contradictory genetic test results or variants of uncertain significance (VUS) were excluded from the cohort, thus only patients carrying known pathogenic variations are discussed in this paper. We excluded two patients from the cohort. One of them had an intronic NF1 variation according to the first NGS, but it was not present with Sanger-sequencing or the repeated NGS from the same DNA sample. This patient was a 6.5-year-old male with 8 CALMs (each was over 5 mm in greatest diameter). Her adult father had 3 CALMs (each was over 15 mm in greatest diameter), but none of the parents had any NF1 variation with NGS and Sanger sequencing. The other excluded female patient had a maternally inherited VUS. This index patient was 4 years old with 7 CALMs (each was over 5 mm in greatest diameter), but her mother did not have any symptoms.
Patients’ classification to survey the utility of NIH criteria
To calculate sensitivity (SE) and specificity (SP) of the NIH diagnostic criteria in childhood, we created four categories of patients according to their clinical classification and NF1 variation status: “clinically diagnosed and genetically confirmed NF1” patients have a pathogenic NF1 variation and fulfill NIH criteria; “clinically not diagnosed but genetically detected NF1” children have a pathogenic NF1 variation but do not fulfill NIH criteria; “clinically diagnosed but genetically not confirmed NF1” patients do not have a NF1 pathogenic variation but fulfill NIH criteria; children with “clinically not diagnosed and genetically not detected NF1” have no pathogenic variation of NF1 and do not fulfill NIH criteria.
SE and SP were calculated according to the following formulas:
Results
NF1-like is a wide, genotypic-phenotypic overlapping spectrum (Table 1)
According to the result of our search many different genetic conditions can mimic the NF1’s appearance, predominantly by their skin manifestations or tissue overgrowth. Certain disorders present real NF1-specific features, while others have similar changes/lesions of a different nature. Most of these entities have a distinct, well-described phenotype, however some might show a very confusing, atypical appearance. Albeit the group of NF1-like syndromes also includes harmless conditions, a great many are linked to cell proliferation/differentiation signaling pathways and are therefore associated with a higher risk for developing tumors. The largest subgroup consists of the RASopathies, which concern the Ras/mitogen-activated protein kinase (RAS/MAPK) network. Most disorders in this class are caused by activating pathogenic variations of different oncogenes that lead to increased tumor development risk in an AD inheritance pattern. Further classes strongly associated with higher tumor risk are the Phosphatase and tensin homolog (PTEN) hamartoma tumor syndromes, the Constitutional mismatch repair deficiency (CMMRD) syndromes, certain types of chromosomal aneuploidies or abnormalities, and the Multiple endocrine neoplasia (MEN) syndromes. PTEN hamartoma tumor syndromes also show an AD inheritance pattern and are characterized by many different localized tumors easily confused with neurofibromas. Fortunately, innocuous conditions can also be found among the NF1-like syndromes, causing only aesthetic lesions. Namely, genetic alterations involving the KIT ligand/c-KIT (KITLG/c-KIT) signaling pathway, which plays an important role in the regulation of skin pigment production, manifest only in pigment changes.
Many NF1-like syndromes show not only phenotypic but also genotypic overlap. Examples of this close relation are Noonan and Lentigines-Electrocardiographic conduction defects-Ocular hypertelorism-Pulmonary stenosis-Abnormalities of the genitalia-Retarded growth-Deafness (LEOPARD) syndromes (both can be caused by Protein Tyrosine Phosphatase Non-Receptor Type 11 gene (PTPN11) pathogenic variation), or the Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha gene (PIK3CA) pathogenic variation related Cowden and Klippel-Trenaunay-Weber syndromes. The NF1-like syndromes’ genetic background and diagnosis are further complicated by the fact that the disease-causing pathogenic variation is not always known (e.g. in 20% of Noonan syndromes [55]).
Clinical characteristics and genetic results of the NF1-like pediatric cohort
Utility of the NIH criteria in childhood (Table 2)
Among 30 children with clinically diagnosed NF1 (P1–16 and P21–34) only 53% (16 patients) had a detectable pathogenic NF1 variation. Ten individuals (P17–20 and P35–40) did not have clinically diagnosed NF1, but four of them (40%) had a detectable pathogenic NF1 pathogenic variation (P17–20). In one further child (P35) additional genetic testing confirmed a PTPN11 pathogenic variation causing Noonan syndrome. In the case of eight genetically NF1 negative patients with CALMs (P29–33 and P38–40) additional Sprouty Related EVH1 Domain Containing 1 gene (SPRED1) analysis excluded the presence of Legius syndrome as well. Overall, the NIH criteria’s sensitivity was 80% (16/20 patients) and its specificity was 30% (6/20 patients) among the examined children.
Components of the NIH criteria (Supplementary material 2)
Each NIH criterion represents the cardinal and/or most common symptoms of NF1. 40% of patients (8/20) with genetically confirmed NF1 had a de novo pathogenic variation. In one of the inherited cases (P19) the affected father did not meet the clinical criteria, he had only 1 CALM. Among children who had a clinical diagnosis of NF1, but tested negative for pathogenic NF1 variations, there were three patients with Legius syndrome (P21–23), two with neurofibromatosis type 2 (NF2) (P24, 25), one with multiple endocrine neoplasia type 2B (MEN2B) (P26), one with LEOPARD syndrome (P27), and one with a 46,XX,inv(4)(p13q13) karyotype (P28). Six individuals without any detected NF1 variation met the NIH criteria (P29–34) - most of them had CALMs and axillary/inguinal freckling -, but none of them had a detectable pathogenic NF1 or SPRED1 variation, or a characteristic phenotype suspicious of any other NF1-like conditions; they should probably have an undetected NF1 variation. Two patients of them have clinically affected family members (P31: mother and brother, P32: mother). In these six cases (P29–34), we intend to perform NF1 and SPRED1 analysis of cultured melanocytes from biopsy of affected skin areas, and to carry out NF1-like gene panel test (including sequencing and copy number variation (CNV) analysis), and in the next step whole exome sequencing (WES) with CNV analysis or whole genome sequencing (WGS) from peripheral blood to exclude the presence of mosaicism or of other NF1-like conditions. Five further children fulfilled only one NIH criterion, and tested negative for an NF1 pathogenic variation (P36–40).
Additional symptoms and anamnestic data (Supplementary material 2)
We collected every additional sign of patients possibly suggestive of NF1 or other NF1-like conditions, excluding those that are known frequent variations in the normal population. For example, 10% of healthy individuals have one or two CALMs, but the presence of three or more is rare (< 1%) and shows a relatively strong association with tumor predisposition syndromes [9]. Although isolated Lisch nodules can also occur in the normal population, they are more common in genetic abnormalities, such as an early sign of germline NF1, the only symptom of segmental NF1, or a rare feature of NF2 [56,57,58]. The same is true for the presence of single neurofibromas.
Occurrence of tumors (Table 3)
In total, 16 (40%) of 40 patients had 21 non-metastatic tumors. Among the different tumors, OPG occurred in the highest number (6/21, 29%): two in patients with verified NF1, one related to NF2, one to chromosome 4 abnormality, and two to NF1 and SPRED1 pathogenic variation double negative cases.
30% (6/20) of children with pathogenic NF1 variation and 50% (10/20) of patients without pathogenic NF1 variation, as well as 27% (8/30) of NIH positive and 30% (3/10) of NIH negative individuals had a minimum of one neoplasm.
Sexes and ages of NF1-like pediatric patients
Based on the ages of patients (Supplementary material 2) we calculated the average time of first genetic counseling in relation to NF1-like syndromes of different patient groups (Fig. 1). Each group is presented with three lines: mean age of the whole patient group, of males and of females.

Average ages at the time of first genetic counseling and evaluation in relation to NF1-like syndromes. Each class is presented with three lines: mean age of the whole group, of males and of females. *: The group includes only patients having detected NF1 pathogenic variant
Among the patients with pathogenic NF1 variation 60% (12 children) were male and 40% (8 children) were female. Half of the 16 patients with clinically diagnosed and genetically confirmed NF1 were males and the other half were females. The group of the clinically not diagnosed but genetically detected NF1 patients included only boys (four patients). Five of the 14 clinically diagnosed but genetically not confirmed NF1 children were females and nine were males. The group of patients without clinically diagnosed and genetically detected NF1 included three females and three males. Half of the patients with pathogenic NF1 variation had a de novo genetic alteration, five of them were male and three were female.
Patient selection for genetic testing of NF1-like syndrome
To support the clinicians in the differential diagnosis, we collected the most extensive spectrum of possible NF1-like syndromes. According to the commonly overlapping phenotype, we recommend for clinicians a detailed, non-invasive clinical evaluation of patients before referring them to a clinical geneticist. Due to the age-related expressivity of most of the NF1-like syndromes, the regular follow-up is also recommended.
Diagnostic and management strategy
Based on the symptoms we could choose the most efficient diagnostic method and the way of patient management. If phenotype and patient history is suspicious but not characteristic, we recommend targeted gene panels (including sequencing and CNV analysis of all NF1-like syndromes’ genes), and in case of a negative result, expansion to WES with CNV analysis or WGS as the most effective diagnostic approach. Here we should mention the diagnostic advantages and limitations of these extensive tests. The targeted gene panel requires less time, has lower cost, reaches higher coverage and sequencing depth than WES/WGS However, WES with CNV analysis or WGS could identify still unknown disease-causing genes, and they might also identify unexpected alterations (e.g. disease-causing repeat number in huntingtin gene) and/or many VUS. WES cannot identify CNVs of a certain size and localization.
In cases for which these tests are inconclusive, but clinical suspicion of NF1-like syndromes remains high, we advocate for ribonucleic acid (RNA)-based or epigenetic tests of suspicious genes, or to consider the possibility of a somatic or undetectable pathogenic variation. In instances of mosaicism the genetic tests might be negative due to the absence of peripheral blood lymphocytes’ involvement. To solve this problem, we should perform the tests on other types of tissues/cells possibly affected by the pathogenic variation (e.g. buccal epithelia). In cases when the genetic background is uncertain, we should consider the possibility of environmental effects or random, rare associations of normal, common phenotypic variants (harmless phenocopy). We should also emphasize that the detection rate of genetic tests - even in combination - do not reach 100%. Therefore, we should take into account the presence of an undetectable pathogenic variation in patients with a clinically diagnosed, but genetically not detected abnormality. Currently there are no methods that can help us clarify these eventualities.
For certain NF1-like syndromes disease-specific monitoring and management guidelines are available and are recommended for genetically confirmed NF1-like syndromes [15].
Discussion
NF1-like syndromes with unclarified tumor predisposing effect
As we have seen on Table 1 the relation between some conditions within this group and tumor predisposition remains unclarified. For instance, Legius syndrome, which is caused by the RAS/MAPK-member SPRED1’s pathogenic variation, behaves as an AD genetic disorder, even though the protein encoded by that gene acts as a tumor suppressor in a homo-, or heterodimer with its paralog Sprouty Related EVH1 Domain Containing 2 (SPRED2). Loss of function pathogenic variations of tumor suppressors are generally considered to have recessive expression patterns and are associated with increased risk of tumor development. However, extensive research over the last few years has elucidated that certain tumor suppressor genes might act as “double agents” with both oncogenic and tumor-suppressor functions. Therefore, in some cases heterozygous pathogenic variations can lead to manifest disease with a dominant inheritance pattern [59]. Legius syndrome is a demonstrative example. SPRED1’s loss of heterozygosity is thought to play a role in pediatric acute myeloblastic leukemia; furthermore, there is some evidence that Legius syndrome is possibly associated with an increased risk for kidney tumors and lung cancers [17,18,19]. Nonetheless, the definitive association between pathogenic SPRED1 variations and tumor predisposition is complicated by the fact that Legius syndrome is very rare, therefore the amount of available evidence is currently insufficient. Whether there is a real increase of certain tumor types in Legius syndrome, or the reported cases represent random association awaits clarification in the future.
Until the significance of these diseases’ tumor predisposing effect remains unclarified, we recommend a regular (yearly) tumor screening/check-ups regarding to the tumors described in the literature. In our cohort, one patient with Legius syndrome (P22) had an epithelioma calcificans, which is a benign tumor and might be a random, independent lesion.
Limitations of the NIH criteria in childhood and genetic testing
The relatively low, 80% sensitivity of the NIH criteria among the examined young patients can be the result of two factors. On the one hand, 46% of sporadic (de novo) cases with NF1 pathogenic variation - which represent nearly half of all individuals with a pathogenic NF1 variation - fail to meet the criteria by 1 year of age because of the absence of an affected first-degree relative. On the other hand, cardinal symptoms of NF1 usually appear in late childhood [11, 15]. Similar to the NIH criteria, genetic testing methods have limitations as well; it is known that there are rare pathogenic variations that are not detectable with genetic tests. In these cases, the NIH criteria could help us in recognition. Two (P31–32) of the six patients with fulfilled NIH criteria but without any detectable NF1 variation (P29–34) probably have a germline variation because of the clinically affected family members.
The 40% prevalence of de novo NF1 pathogenic variations in our cohort approaches the 50% rate known from the scientific literature that is typical for monogenic disorders with an AD inheritance pattern and age-related expressivity. 75% (6/8) of presented patients with sporadic NF1 fulfilled the criteria, which is higher than the expected 46% [11]. However, DeBella and her colleagues’ results referred to children under 1 year of age, while our cohort included older patients.
Explaining the NIH criteria’s low specificity in the examined children is more complex. By reviewing the patients’ symptoms, we can see that several NF1-like conditions may also present with six or more CALMs, axillary and/or inguinal freckling, at least one clinically affected family member, and sometimes even neurofibromas and Lisch nodules. This shows that these criteria are the commonest, but not the most specific symptoms of NF1.
In the presented pediatric cohort NF1-like syndromes affected an approximately equal number of males and females, which was also true in the subgroup of patients with NF1 pathogenic variation. The near equivalent efficacy of the NIH criteria in different sexes suggests that the prevalence and age-related appearance of the most cardinal NF1 symptoms are independent from gender. As there was a significant difference in the average age of patients with clinically diagnosed and genetically confirmed NF1 vs. children with clinically not diagnosed but genetically detected NF1 (10.5 vs. 5 years), the NIH criteria’s utility seems to be limited in early years of life. The difference between the mean ages of children with de novo and inherited NF1 pathogenic variations (5 vs. 12 years) could be due to parental ascertainment bias: new and unknown symptoms in a family are more likely to cause concern than signs which are already present in family members. These latter familial, and usually painless symptoms are often perceived to be normal variants, and do not tend to urge parents to seek medical help.
The average age of patients having a tumor was 9 years, which highlights the need for NF1-like syndromes’ early diagnosis. The relatively high average age in MEN2B, LEOPARD and Noonan syndromes further represents the diagnostic challenge of these rare genetic disorders. In contrast, a chromosomal rearrangement can be discernible earlier due to frequent addition of congenital anomalies, which was illustrated by the case of a two-year-old patient with 46,XX,inv(4)(p13q13) karyotype.
As we have seen in a patient (P19) with inherited NF1 pathogenic variation, the affected father did not meet the clinical diagnostic criteria. His case is instructive for many reasons. Firstly, we should note that there are rare cases in which the NIH criteria are not fulfilled despite entering adulthood. This phenomenon could be explained by NF1’s variable expressivity, the presence of mosaicism, or a type of pathogenic variation that causes only skin pigment lesions. Secondly, inheritance should not be based solely on clinical findings, genetic testing is always necessary to ascertain inheritance. Finally, the phenomenon of germline/gonadal mosaicism can explain uncertain heritability [60]: mosaicism of germinal cells can lead to apparently de novo pathogenic variations in the offspring, similar to some cases, which have been reported so far [61,62,63,64]. A further point of interest from the scientific literature is the observation of different NF1, or simultaneous NF1 and SPRED1 pathogenic variations within one family each [65], which can be attributed to NF1’s high variation rate and/or the phenomenon that the presence of one variation predisposes for another.
Clinical relevance of additional symptoms and tumors in the NF1-like spectrum
NF1 is also associated with many characteristic features other than components of the NIH criteria (e.g. short stature, macrocephaly, unidentified bright objects (UBOs) on brain MRI, etc.), which can also present as first manifestations, thus young patients showing exclusively these symptoms do not receive the correct diagnosis, necessary tumor surveillance and therapy in time. Some of these features are very specific for NF1 but can be more difficult to diagnose (e.g. ascertaining the presence of UBOs requires an MRI, which necessitates anesthesia in young children). Naturally, this does not mean that we suggest performing routine brain scans in every suspected case indiscriminately, rather we propose that some of the additional symptoms should be considered when establishing clinical diagnostic criteria for NF1. UBOs are thought to have a weak relation with cognitive impairment, which is attributed to the possible presence of aberrant myelination or gliosis [15]. The tenuous link between UBOs and cognitive impairment is further weakened by our findings, i.e. that none of the patients with intellectual impairment had UBOs, and vice versa.
40% (16/40) of the cohort had at least one tumor and 10% (4/40) had two or more, which demonstrates the significant tumor predisposition effect of the entire NF1-spectrum. 30% (6/20) of the patients with genetically confirmed NF1 had minimum one tumor, 10% (2/20) of which were OPG, and this prevalence approaches the 15–20% cited in the scientific literature [66]. While children with NF1 have an increased risk for developing low-grade optic pathway and brainstem gliomas, the NF2 typically associates with low-grade tumors of cranial nerves (vestibular schwannomas), meninges (meningiomas) and spinal cord (ependymomas) [67]. This association is illustrated by two patients: patient (P20) with NF1 had an astrocytoma in the brain stem, and patient P25 with NF2 had a meningioma and bilateral acoustic neuromas. Although OPGs are thought to be usually specific to NF1, they were also found in other members of our cohort namely in one patient with NF2 (P24), one with the chromosome 4 rearrangement (P28), and two children without any detected pathogenic NF1 or SPRED1 variation (P29 and P34) (Table 3). These cases could be explained by the presence of mosaic NF1 pathogenic variation, epigenetic alterations affecting NF1’s expression or the effect of its protein, as well as the association of this tumor type with other genetic diseases, or random, unfortunate association of sporadic OPG. Diagnosis of OPG with CT/MRI is possible in cases with characteristic imaging features, such as iso- or hyper-intense lesions on CT that usually enhance after contrast injection, or hypo- to iso-intense appearance on T1 MRI, and hyperintensity on T2 images. Biopsy of the suspicious lesion is offered to confirm the diagnosis only in patients with unusual clinical or imaging findings [68,69,70,71,72]. All OPG in our cohort showed characteristic appearance on MRI, therefore biopsy was not performed in our patients to avoid redundant, severe complications. The OPG of P24 with genetically detected pathogenic NF2 variation was diagnosed neuroradiologically because of the OPG-characteristic appearance on MRI, therefore no biopsy from the tumor was performed.
After OPG, acoustic neuroma was the second most frequent tumor among the presented patients. At a young patient (P6) a unilateral ductal carcinoma of breast was detected. Higher incidence of breast cancer in NF1 is likewise known from the scientific literature, but its emergence in childhood is very rare. One patient with NF1 pathogenic variation (P2) had pituitary adenoma that can cause further subsequent symptoms depending on its hormone production. Juvenile myelomonocytic leukemia (JMML) can also be a severe complication of NF1 [73], but did not occur in this cohort. The patient P36 had two plexiform neurofibromas and many other types of tumors in one area of the body, but there was no detectable NF1 pathogenic variation in DNA isolated from peripheral blood sample, which was suggestive for segmental NF1. The abovementioned heterogeneity and development of tumors in NF1 can be attributed to the Knudson „two-hit” model. According to this concept, loss of heterozygosity is necessary for the development of benign tumors, especially neurofibromas, but not enough for the formation of malignancies. Evidence exists that further genetic aberration is required for malignant transformation in NF1 [74].
Suggestions to improve the diagnostic efficiency of NF1-like syndromes in childhood
Considering the broad spectrum and diagnostic difficulties of overlapping diseases, we propose the comprehensive use of NF1-like conditions/syndromes, including NF1 as well as disorders with phenocopy of NF1. We emphasize the importance of an exhaustive clinical examination, family tree analysis and anamnesis, which essentially support the decisions regarding appropriate diagnostics and management. To guide physicians in the systematic assessment of a patient’s symptoms, and to assist in judging whether a child requires genetic evaluation, we provide a more detailed, non-invasive clinical evaluation of patients before referring them to a clinical geneticist.
Management of patients with mosaic or clinically diagnosed but genetically undetectable NF1
As we cannot test all tissue types for NF1 pathogenic variations, we do not know the extent of affected tissues. Therefore, patients with mosaic NF1 should be examined in the beginning and regularly followed-up until entering adulthood with the non-invasive tests also recommended for patients with non-mosaic NF1. Furthermore, patients with clinically diagnosed but genetically undetectable NF1 should be also regularly checked-up because of the high probability of cryptic pathogenic NF1 variation and risk for NF1-related complications.
Limitations and future perspectives
Unfortunately, it was not always possible to accurately register the time of the symptoms’ first appearance, due to the young age of the patients, parental uncertainty and/or lack of exact medical documentation. In most cases, the parents bring their children to the genetic counseling because of CALMs, which are commonly the first manifestation of NF1. Beside parents, the pediatricians also play an important role in recognition of NF1-like syndromes’ initial features. In Hungary the children are mandatorily checked-up at specific dates by well-trained home pediatricians. The precise registration of first presentation might be improved by careful patient and family education, which we plan to implement in the future.
The unusually high number of non NF1-related tumors could be the consequence of our clinic’s main profiles, one of which is pediatric tumors. Although the genetic division provides for patients with various genetic diseases (e.g. developmental delay, metabolic diseases, chromosomal abnormalities, congenital disorders, etc.), the number of children diagnosed with a tumor is significant. As the patients to this study were collected because of a wider spectrum of NF1-like syndromes and features, and consequently underwent NF1’s genetic testing, the cohort and our results could have a bias of more and mostly pediatric tumor related NF1-like syndromes. Although the number of patients managed by our clinic is limited, our results broaden the observations and support the knowledge about NF1 and other NF1-like conditions.
The diagnostic difficulties of NF1 have been discussed in the recent years and has led to an international consortium that revised the diagnostic criteria: the NIH criteria were retained with some changes and supplementary new elements (the recommendations are yet unpublished) [75,76,77,78]. According to this the following changes have been recommended:
-
axillary or inguinal freckling should be bilateral;
-
ophthalmic alterations could be not only ≥2 Lisch nodules, but also ≥2 choroidal abnormalities;
-
distinctive osseus lesion could be not only sphenoid wing dysplasia, but also anterolateral bowing of tibia (tibial dysplasia), or pseudarthrosis of a long bone;
-
the affected first degree relative with clinically diagnosed NF1 should be a parent;
-
a new diagnostic element is the presence of an NF1 heterozygous pathogenic variant.
According to the revised criteria, the classification of three patients’ cases in our cohort have been changed: P17, P18 and P20 fulfilled the new NF1 diagnostic criteria. Therefore, the sensitivity of the revised criteria was higher (93%) than according to the NIH criteria (80%), and the specificity did not decline (30%).
Our results emphasize the need for widespread use of the revised criteria, with the help of which the diagnostic effectivity of NF1 could be improved.
Conclusions
We propose the consistent use of the term NF1-like syndromes for conditions showing NF1-like appearance, that may be useful in cases without a clarified genetic background, whereby we can shelter the patients from an unnecessary stigmatizing diagnosis and simultaneously emphasize the need of regular medical follow-up until adulthood. Due to the poor manifestation of NF1-like syndromes in childhood, we propose careful appraisal of additional signs and symptoms other than the NIH criteria and recommend yearly clinical reevaluation to detect newly emerging symptoms and to follow-up the patient’s development.
Availability of data and materials
Not applicable.
Abbreviations
- ACTB/G1 :
-
Actin Beta/Gamma 1 gene
- AD:
-
Autosomal dominant
- AKT1 :
-
AKT Serine/Threonine Kinase 1 gene
- ANAPC1 :
-
Anaphase Promoting Complex Subunit 1 gene
- AR:
-
Autosomal recessive
- ATM :
-
ATM Serine/Threonine Kinase gene
- BRAF :
-
B-Raf Proto-Oncogene, Serine/Threonine Kinase gene
- BRCA1/2 :
-
BRCA1/2 DNA Repair Associated gene
- BRIP1 :
-
BRCA1 Interacting Protein C-Terminal Helicase 1 gene
- CALM(s):
-
Café-au-lait macule(s)
- CBL sy.:
-
CBL gene mutation syndrome
- CBL :
-
Cbl Proto-Oncogene gene
- CCND1 :
-
Cyclin D1 gene
- CDKN1C :
-
Cyclin Dependent Kinase Inhibitor 1C gene
- chr:
-
Chromosome
- CMMRD:
-
Constitutional mismatch repair deficiency
- CNV(s):
-
Copy number variation(s)
- CREBBP :
-
CREB Binding Protein gene
- del:
-
Deletion
- DNA:
-
Deoxyribonucleic acid
- dup:
-
Duplication
- EP300 :
-
E1A Binding Protein P300 gene
- EPHB4 :
-
EPH Receptor B4 gene
- ERCC4 :
-
ERCC Excision Repair 4, Endonuclease Catalytic Subunit gene
- FANCA/B/C/D2/E/F/I/L :
-
FA Complementation Group A/B/C/D2/E/F/I/L gene
- FBN1 :
-
Fibrillin 1 gene
- FISH:
-
Fluorescent in situ hybridization
- GBA :
-
Glucosylceramidase Beta gene
- GNAS :
-
GNAS Complex Locus gene
- HMGA2 :
-
High Mobility Group AT-Hook 2 gene
- HRAS :
-
HRas Proto-Oncogene, GTPase gene
- ICR1:
-
Imprinting control region 1
- IFNG :
-
Interferon Gamma gene
- IGF2 :
-
Insulin Like Growth Factor 2 gene
- KDM6A :
-
Lysine Demethylase 6A gene
- KIT :
-
KIT Proto-Oncogene, Receptor Tyrosine Kinase gene
- KITLG :
-
KIT Ligand gene
- KITLG/c-KIT:
-
KIT ligand/c-KIT
- KLLN :
-
Killin, P53 Regulated DNA Replication Inhibitor gene
- KMT2D :
-
Lysine Methyltransferase 2D gene
- KRAS :
-
KRAS Proto-Oncogene, GTPase gene
- LEOPARD:
-
Lentigines-Electrocardiographic conduction defects-Ocular hypertelorism-Pulmonary stenosis-Abnormalities of the genitalia-Retarded growth-Deafness syndrome
- LZTR1 :
-
Leucine Zipper Like Transcription Regulator 1 gene
- MAD2L2 :
-
Mitotic Arrest Deficient 2 Like 2 gene
- MAP 2 K1/2 :
-
Mitogen-Activated Protein Kinase Kinase 1 gene
- MEN:
-
Multiple endocrine neoplasia
- MEN1 :
-
Menin 1 gene
- MLH1 :
-
MutL Homolog 1 gene
- MLPA:
-
Multiplex ligation dependent probe amplification
- MRAS :
-
Muscle RAS Oncogene Homolog gene
- MRI:
-
Magnetic resonance imaging
- MSH2/6 :
-
MutS Homolog 2/6 gene
- NBN :
-
Nibrin gene
- NF1:
-
Neurofibromatosis type 1
- NF1 :
-
Neurofibromin 1 gene
- NF2:
-
Neurofibromatosis type 2
- NF2 :
-
Neurofibromin 2 gene
- NGS:
-
Next-generation sequencing
- NIH:
-
National Institutes of Health (U.S.)
- NRAS :
-
NRAS Proto-Oncogene, GTPase gene
- OMIM:
-
Online Mendelian Inheritance in Man
- OMIM ID:
-
Online Mendelian Inheritance in Man Identifier
- OMIM PS:
-
Phenotypic Series in Online Mendelian Inheritance in Man (similar phenotypes with heterogeneous genetic background)
- OPG:
-
Optic pathway glioma
- ORPHA ID:
-
Orphanet: an online database of rare diseases and orphan drugs Identifier
- p:
-
Short arm of a chromosome
- P:
-
Patient
- PALB2 :
-
Partner And Localizer Of BRCA2 gene
- PCNT :
-
Pericentrin gene
- PI3K:
-
Phosphoinositide-3-kinase-protein kinase
- PIK3CA :
-
Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha gene
- PLAG1 :
-
PLAG1 Zinc Finger gene
- PMS2 :
-
PMS1 Homolog 2, Mismatch Repair System Component gene
- PPP1CB :
-
Protein Phosphatase 1 Catalytic Subunit Beta gene
- PRKAR1A :
-
Protein Kinase CAMP-Dependent Type I Regulatory Subunit Alpha gene
- PTCH1/2 :
-
Patched 1/2 gene
- PTEN:
-
Phosphatase and tensin homolog
- PTEN :
-
Phosphatase and tensin homolog gene
- PTPN11 :
-
Protein Tyrosine Phosphatase Non-Receptor Type 11 gene
- RAD51 :
-
RAD51 Recombinase gene
- RAD51C :
-
RAD51 Paralog C gene
- RAF1 :
-
Raf-1 Proto-Oncogene, Serine/Threonine Kinase gene
- RAS/MAPK:
-
Ras/mitogen-activated protein kinase
- RASA1 :
-
RAS P21 Protein Activator 1 gene
- RECQL3/4 :
-
RecQ Like Helicase 3/4 gene
- RET :
-
Ret Proto-Oncogene gene
- RFWD3 :
-
Ring Finger And WD Repeat Domain 3 gene
- RIT1 :
-
Ras Like Without CAAX 1 gene
- RNA:
-
Ribonucleic acid
- RRAS2 :
-
RAS Related 2 gene
- SE:
-
Sensitivity
- SEC23B :
-
SEC23 Homolog B, COPII Coat Complex Component gene
- SHOC2 :
-
SHOC2 Leucine Rich Repeat Scaffold Protein gene
- SLX4 :
-
SLX4 Structure-Specific Endonuclease Subunit gene
- SMARCB1 :
-
SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily B, Member 1 gene
- SNAI2 :
-
Snail Family Transcriptional Repressor 2 gene
- SOS1/2 :
-
SOS Ras/Rac Guanine Nucleotide Exchange Factor 1/2 gene
- SP:
-
Specificity
- SPRED1/2 :
-
Sprouty Related EVH1 Domain Containing 1/2 gene
- SPRED2:
-
Sprouty Related EVH1 Domain Containing 2 protein
- STK11 :
-
Serine/Threonine Kinase 11 gene
- SUFU :
-
SUFU Negative Regulator Of Hedgehog Signaling gene
- sy(s).:
-
Syndrome(s)
- t:
-
Translocation
- TOP3A :
-
DNA Topoisomerase III Alpha gene
- TSC1/2 :
-
TSC Complex Subunit 1/2 gene
- UBE2T :
-
Ubiquitin Conjugating Enzyme E2 T gene
- UBO(s):
-
Unidentified bright object(s)
- UBR1 :
-
Ubiquitin Protein Ligase E3 Component N-Recognin 1 gene
- UPD:
-
Uniparental disomy
- VHL :
-
Von Hippel-Lindau Tumor Suppressor gene
- VUS:
-
Variant of uncertain significance
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
- XRCC2/9 :
-
X-Ray Repair Cross Complementing 2/9 gene
References
Legius E. Neurofibromatosis type 1. Orphanet encyclopedia, 2014, https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=636.
Radtke HB, Sebold CD, Allison C, Haidle JL, Schneider G. Neurofibromatosis type 1 in genetic counseling practice: recommendations of the National Society of genetic counselors. J Genet Couns. 2007;16(4):387–407. https://doi.org/10.1007/s10897-007-9101-8.
Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1–16. https://doi.org/10.1016/j.jaad.2008.12.051.
NIH. National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md, USA, July 13–15, 1987. Neurofibromatosis. 1988;1:172–8.
Friedman JM. Neurofibromatosis 1. 1998 Oct 2 [updated 2019 Jun 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1109/.
Kresak JL, Walsh M. Neurofibromatosis: a review of NF1, NF2, and Schwannomatosis. J Pediatr Genet. 2016;5(2):98–104. https://doi.org/10.1055/s-0036-1579766.
Shah KN. The diagnostic and clinical significance of café-au-lait macules. Pediatr Clin N Am. 2010;57(5):1131–53. https://doi.org/10.1016/j.pcl.2010.07.002.
Zhang J, Li M, Yao Z. Molecular screening strategies for NF1-like syndromes with café-au-lait macules. Mol Med Rep. 2016;14(5):4023–9. https://doi.org/10.3892/mmr.2016.5760.
Santos AC, Heck B, Camargo BD, Vargas FR. Prevalence of Café-au-Lait spots in children with solid tumors. Genet Mol Biol. 2016;39(2):232–8. https://doi.org/10.1590/1678-4685-GMB-2015-0024.
Jongmans MCJ, Loeffen JLCM, Waanders E, Hoogerbrugge PM, Ligtenberg MJL, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: an easy-to-use selection tool. Eur J Med Genet. 2016;59(3):116–25. https://doi.org/10.1016/j.ejmg.2016.01.008.
DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of Neurofibromatosis 1 in children. Pediatrics. 2000;105(3):608–14. https://doi.org/10.1542/peds.105.3.608.
Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD), 26th March 2020. World Wide Web URL:https://omim.org/.
Orphanet: an online database of rare diseases and orphan drugs. Copyright, INSERM 1997. Available at http://www.orpha.net. Accessed 26 Mar 2020.
Ly KI, Blakeley O. The diagnosis and Management of Neurofibromatosis Type 1. Med Clin North Am. 2019;103(6):1035–54. https://doi.org/10.1016/j.mcna.2019.07.004.
Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81–8. https://doi.org/10.1136/jmg.2006.045906.
Stevenson D, Viskochil D, Mao R. Legius syndrome. 2010 Oct 14 [updated 2015 Jan 15]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47312/.
Pasmant E, Gilbert-Dussardier B, Petit A, de Laval B, Luscan A, Gruber A, et al. SPRED1, a RAS MAPK pathway inhibitor that causes Legius syndrome, is a tumour suppressor downregulated in paediatric acute myeloblastic leukaemia. Oncogene. 2015;34(5):631–8. https://doi.org/10.1038/onc.2013.587.
Sun J, Zhang J, Wang Y, Li Y, Zhang R. A pilot study of aberrant CpG Island Hypermethylation of SPRED1 in acute Myeloloid leukemia. Int J Med Sci. 2019;16(2):324–30. https://doi.org/10.7150/ijms.27757.
Brems H, Legius E. Legius syndrome, an update. Molecular pathology of mutations in SPRED1. Keio J Med. 2013;62(4):107–12. https://doi.org/10.2302/kjm.2013-0002-RE.
Jongmans MCJ, van der Burgt I, Hoogerbrugge PM, Noordam K, Yntema HG, Nillesen WM, et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet. 2011;19(8):870–4. https://doi.org/10.1038/ejhg.2011.37.
Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, Cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157(2):83–9. https://doi.org/10.1002/ajmg.c.30300.
Garavelli L, Cordeddu V, Errico S, Bertolini P, Street ME, Rosato S, et al. Noonan syndrome-like disorder with loose anagen hair: a second case with neuroblastoma. Am J Med Genet A. 2015;167A(8):1902–7. https://doi.org/10.1002/ajmg.a.37082.
Huckstadt V, Chinton J, Gomez A, Obregon MG, Gravina LP. Noonan syndrome with loose anagen hair with variants in the PPP1CB gene: first familial case reported. Am J Med Genet A. 2021;185(4):1256–60. https://doi.org/10.1002/ajmg.a.62089.
Niemeyer CM. JMML genomics and decisions. Hematology Am Soc Hematol Educ Program. 2018;2018(1):307–12. https://doi.org/10.1182/asheducation-2018.1.307.
Bayrak-Toydemir P, Stevenson D. Capillary malformation-arteriovenous malformation syndrome. 2011 Feb 22 [updated 2019 Sep 12]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK52764/.
Piqueres-Zubiaurre T, Martínez de Lagrán Z, González-Pérez R, Urtaran-Ibarzabal A, Perez de Nanclares G. Familial progressive hyperpigmentation, cutaneous Mastocytosis, and gastrointestinal stromal tumor as clinical manifestations of mutations in the c-KIT receptor gene. Pediatr Dermatol. 2017;34(1):84–9. https://doi.org/10.1111/pde.13040.
Cohen MM. Proteus syndrome review: molecular, clinical, and pathologic features. Clin Genet. 2014;85(2):111–9. https://doi.org/10.1111/cge.12266.
Plevová P. An Update on Inherited Colon Cancer and Gastrointestinal Polyposis. Klin Onkol. 2019;32(Supplementum2):97–108. https://doi.org/10.14735/amko2019S97.
Bhargava R, Au Yong KJ, Leonard N. Bannayan-Riley-Ruvalcaba syndrome: MRI neuroimaging features in a series of 7 patients. AJNR Am J Neuroradiol. 2014;35(2):402–6. https://doi.org/10.3174/ajnr.A3680.
Macken WL, Tischkowitz M, Lachlan KL. PTEN hamartoma tumor syndrome in childhood: a review of the clinical literature. Am J Med Genet C Semin Med Genet. 2019;181(4):591–610. https://doi.org/10.1002/ajmg.c.31743 [Epub ahead of print].
van Os NJH, Haaxma CA, van der Flier M, Merkus PJFM, van Deuren M, de Groot IJM, et al. Ataxia-telangiectasia: recommendations for multidisciplinary treatment. Dev Med Child Neurol. 2017;59(7):680–9. https://doi.org/10.1111/dmcn.13424.
Diets IJ, Waanders E, Ligtenberg MJ, van Bladel DAG, Kamping EJ, Hoogerbrugge PM, et al. High yield of pathogenic germline mutations causative or likely causative of the Cancer phenotype in selected children with Cancer. Clin Cancer Res. 2018;24(7):1594–603. https://doi.org/10.1158/1078-0432.CCR-17-1725.
Latham SL, Ehmke N, Reinke PYA, Taft MH, Eicke D, Reindl T, et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat Commun. 2018;9(1):4250. https://doi.org/10.1038/s41467-018-06713-0.
Cianci P, Fazio G, Casagranda S, Spinelli M, Rizzari C, Cazzaniga G, et al. Acute myeloid leukemia in Baraitser-winter cerebrofrontofacial syndrome. Am J Med Genet A. 2017;173(2):546–9. https://doi.org/10.1002/ajmg.a.38057.
Campbell MB, Campbell WC, Rogers J, Rogers N, Rogers Z, van den Hurk AM, et al. Bloom syndrome: research and data priorities for the development of precision medicine as identified by some affected families. Cold Spring Harb Mol Case Stud. 2018;4(2):a002816. https://doi.org/10.1101/mcs.a002816.
Kamilaris CDC, Faucz FR, Voutetakis A, Stratakis CA. Carney complex. Exp Clin Endocrinol Diabetes. 2019;127(2–03):156–64. https://doi.org/10.1055/a-0753-4943.
Nepal M, Che R, Zhang J, Ma C, Fei P. Fanconi Anemia signaling and Cancer. Trends Cancer. 2017;3(12):840–56. https://doi.org/10.1016/j.trecan.2017.10.005.
Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017;18(2):E441. https://doi.org/10.3390/ijms18020441.
Hasan A, Akintola D. An update of Gorlin-Goltz syndrome. Prim Dent J. 2018;7(3):38–41. https://doi.org/10.1177/205016841800700306.
Hoffman WH, Lee JR, Kovacs K, Chen H, Yaghmai F. Johanson-blizzard syndrome: autopsy findings with special emphasis on hypopituitarism and review of the literature. Pediatr Dev Pathol. 2007;10(1):55–60. https://doi.org/10.2350/06-05-0085.1.
Scala M, Morana G, Sementa AR, Merla G, Piatelli G, Capra V, et al. Aggressive desmoid fibromatosis in kabuki syndrome: expanding the tumor spectrum. Pediatr Blood Cancer. 2019;66(9):e27831. https://doi.org/10.1002/pbc.27831.
Edmondson AC, Kalish JM. Overgrowth syndromes. J Pediatr Genet. 2015;4(3):136–43. https://doi.org/10.1055/s-0035-1564440.
Dietz H. Marfan syndrome. 2001 Apr 18 [updated 2017 Oct 12]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1335/.
Boyce AM, Florenzano P, de Castro LF, Collins MT. Fibrous dysplasia/McCune-Albright syndrome. 2015 Feb 26 [updated 2019 Jun 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK274564/.
Decmann A, Patócs A, Igaz P. Overview of genetically determined diseases/multiple endocrine neoplasia syndromes predisposing to endocrine tumors. Exp Suppl. 2019;111:105–27. https://doi.org/10.1007/978-3-030-25905-1_7.
Bober MB, Jackson AP. Microcephalic Osteodysplastic primordial dwarfism, Type II: a Clinical Review. Curr Osteoporos Rep. 2017;15(2):61–9. https://doi.org/10.1007/s11914-017-0348-1.
Silveri M, Grossi A, Bassani F, Orazi C, Camassei FD, Zaccara A. Ullrich-turner syndrome and tumor risk: is there another chance to early Gonadectomy in positive TSPY and SRY patients? Eur J Pediatr Surg. 2016;26(3):273–6. https://doi.org/10.1055/s-0035-1551568.
Ma M, Ding ZL, Cheng ZQ, Wu G, Tang XY, Deng P, et al. Neurocutaneous melanosis in an adult patient with intracranial primary malignant melanoma: case report and review of the literature. World Neurosurg. 2018;114:76–83. https://doi.org/10.1016/j.wneu.2018.02.007.
Varon R, Demuth I, Chrzanowska KH. Nijmegen breakage syndrome. 1999 may 17 [updated 2017 Feb 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1176/.
Marsh D. Proteus-like syndrome. Orphanet encyclopedia, 2013, https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=2969.
Boot MV, van Belzen MJ, Overbeek LI, Hijmering N, Mendeville M, Waisfisz Q, et al. Benign and malignant tumors in Rubinstein-Taybi syndrome. Am J Med Genet A. 2018;176(3):597–608. https://doi.org/10.1002/ajmg.a.38603.
Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM, Salem J, Bliek J, et al. Diagnosis and management of silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13(2):105–24. https://doi.org/10.1038/nrendo.2016.138.
Lam HC, Nijmeh J, Henske EP. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J Pathol. 2017;241(2):219–25. https://doi.org/10.1002/path.4827.
Chou A, Toon C, Pickett J, Gill AJ. Von Hippel-Lindau syndrome. Front Horm Res. 2013;41:30–49. https://doi.org/10.1159/000345668.
Bhambhani V, Muenke M. Noonan syndrome. Am Fam Physician. 2014;89(1):37–43.
Ceuterick SD, Van Den Ende JJ, Smets RME. Clinical and genetic significance of unilateral Lisch nodules. Bull Soc Belge Ophtalmol. 2005;295:49–53.
Senthilkumar VA, Tripathy K. Lisch Nodules. 2021 Feb 14. In: StatPearls. Treasure Island: StatPearls Publishing; 2021. PMID: 32491492.
Adams EG, Stewart KMA, Borges OA, Darling T. Multiple, unilateral lisch nodules in the absence of other manifestations of neurofibromatosis type 1. Case Rep Ophthalmol Med. 2011;2011:854784–2. https://doi.org/10.1155/2011/854784.
Datta N, Chakraborty S, Basu M, Ghosh MK. Tumor suppressors having oncogenic functions: the double agents. Cells. 2020;10(1):46. https://doi.org/10.3390/cells10010046.
Zlotogora J. Germ line mosaicism. Hum Genet. 1998;102(4):381–6. https://doi.org/10.1007/s004390050708.
Trevisson E, Forzan M, Salviati L, Clementi M. Neurofibromatosis type 1 in two siblings due to maternal germline mosaicism. Clin Genet. 2014;85(4):386–9. https://doi.org/10.1111/cge.12177.
Muram-Zborovski TM, Vaughn CP, Viskochil DH, Hanson H, Mao R, Stevenson DA. NF1 exon 22 analysis of individuals with the clinical diagnosis of neurofibromatosis type 1. Am J Med Genet A. 2010;152A(8):1973–8. https://doi.org/10.1002/ajmg.a.33525.
Bottillo I, Torrente I, Lanari V, Pinna V, Giustini S, Divona L, et al. Germline mosaicism in neurofibromatosis type 1 due to a paternally derived multi-exon deletion. Am J Med Genet A. 2010;152A(6):1467–73. https://doi.org/10.1002/ajmg.a.33386.
Lázaro C, Gaona A, Lynch M, Kruyer H, Ravella A, Estivill X. Molecular characterization of the breakpoints of a 12-kb deletion in the NF1 gene in a family showing germ-line mosaicism. Am J Hum Genet. 1995;57(5):1044–9.
Pacot L, Burin des Roziers C, Laurendeau I, Briand-Suleau A, Coustier A, Mayard T, et al. One NF1 Mutation may Conceal Another. Genes (Basel). 2019;10(9):E633. https://doi.org/10.3390/genes10090633.
Campen CJ, Gutmann DH. Optic pathway gliomas in Neurofibromatosis type I. J Child Neurol. 2018;33(1):73–81. https://doi.org/10.1177/0883073817739509.
Campian J, Gutmann DH. CNS tumors in Neurofibromatosis. J Clin Oncol. 2017;35(21):2378–85. https://doi.org/10.1200/JCO.2016.71.7199.
Huang M, Patel J, Patel BC. Optic Nerve Glioma. 2021 Feb 25. In: StatPearls. Treasure Island: StatPearls Publishing; 2021. PMID: 32491801.
Fried I, Tabori U, Tihan T, Reginald A, Bouffet E. Optic pathway gliomas: a review. CNS Oncol. 2013;2(2):143–59. https://doi.org/10.2217/cns.12.47.
Binning MJ, Liu JK, Kestle JRW, Brockmeyer DL, Walker ML. Optic pathway gliomas: a review. Neurosurg Focus. 2007;23(5):E2. https://doi.org/10.3171/FOC-07/11/E2.
Singhal S, Birch JM, Kerr B, Lashford L, Evans DGR. Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child. 2002;87(1):65–70. https://doi.org/10.1136/adc.87.1.65.
Papini C, Dineen RA, Walker DA, Thomas S, Pitchford NJ. Neuropsychological outcomes of children with optic pathway glioma. Sci Rep. 2020;10(1):3344. https://doi.org/10.1038/s41598-020-59896-2.
Niemeyer CM, Flotho C. Juvenile myelomonocytic leukemia: who’s the driver at the wheel? Blood. 2019;133(10):1060–70. https://doi.org/10.1182/blood-2018-11-844688.
Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26(32):4609–16. https://doi.org/10.1038/sj.onc.1210261.
Rosser T. Neurocutaneous Disorders. Continuum (Minneap Minn). 2018;24(1, Child Neurology):96–129. https://doi.org/10.1212/CON.0000000000000562.
Ruggieri M, Praticò AD. Mosaic Neurocutaneous disorders and their causes. Semin Pediatr Neurol. 2015;22(4):207–33. https://doi.org/10.1016/j.spen.2015.11.001.
Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14(1):355–69. https://doi.org/10.1146/annurev-genom-091212-153523.
Anderson S. Café au Lait macules and associated genetic syndromes. J Pediatr Health Care. 2020;34(1):71–81. https://doi.org/10.1016/j.pedhc.2019.05.001.
Acknowledgements
The authors would like to thank the patients and parents/legal guardians for their collaboration, and the Department of Medical Genetics, University of Pécs for carrying out a substantial part of molecular genetic tests.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. (All genetic tests were performed with permission of the National Health Insurance Fund of Hungary in every individual case.)
Author information
Authors and Affiliations
Contributions
EP analyzed and interpreted the patients’ data, reviewed the literature and was a major contributor in writing the manuscript. KN and KS isolated the DNA from the patients’ peripheral blood samples and implemented most of the molecular genetic tests. (Other genetic tests were performed at further national and international laboratories with permission of the National Health Insurance Fund of Hungary in every individual case.) IH made and analyzed the cytogenetic tests and contributed in writing the manuscript. GyF performed the physical examination and medical check-ups of the patients and the genetic counseling for their parents/legal guardians. AL checked the language grammar and contributed in writing the manuscript. All authors read, checked and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
None of the applied tests, examinations or protocols were experimental. All tests, examinations and protocols were diagnostic, were approved by the institutional committee and were in accordance to guidelines of national, international, institutional and Declaration of Helsinki in the manuscript. A written informed consent has been obtained from the participants/parents/legal guardians to participate in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary material 1.
NF1 variation testing of the cohort. All 40 patients were tested for NF1 pathogenic variation with combination of cyto- and molecular genetic methods.
Additional file 2: Supplementary material 2.
Symptoms and data of the cohort. All patients were tested for NF1, but some also for other suspicious NF1-like genes based on the main symptoms. Fulfilled NIH criteria are highlighted in bold. *: The parent had clinical diagnosis confirmed by genetic test. Abbreviations: IUGR: intrauterine growth retardation.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pinti, E., Nemeth, K., Staub, K. et al. Diagnostic difficulties and possibilities of NF1-like syndromes in childhood. BMC Pediatr 21, 331 (2021). https://doi.org/10.1186/s12887-021-02791-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-021-02791-0