
Abstract
Background
Li-Fraumeni syndrome (LFS), a hereditary condition attributed to TP53 pathogenic variants,(PV), is associated with high risks for various malignant tumors, including breast cancer. Notably, individuals harboring TP53 PVs are more likely (67–83%) to develop HER2 + breast cancer than noncarriers (16–25%). In this retrospective study, we evaluated the associations between TP53 variants and breast cancer phenotype.
Methods
We conducted a retrospective review of the medical records of patients with LFS treated at a single institution and reviewed the literature on TP53 functions and the mechanisms underlying HER2 + breast cancer development in LFS.
Results
We analyzed data for 10 patients with LFS from 8 families. The median age at the onset of the first tumor was 35.5 years. Only case 2 met the classic criteria; this patient harbored a nonsense variant, whereas the other patients carried missense variants. We observed that 9 of 10 patients developed breast cancer. Immunohistochemical analyses revealed that 40% of breast cancers in patients with LFS were HR − /HER2 + . The median age at the onset of breast cancer was slightly younger in HR − /HER2 + tumors than in HR + /HER2 − tumors (31 years and 35.5 years, respectively).
Conclusions
The occurrence of HER2 + breast cancer subtype was 40% in our LFS case series, which is greater than that in the general population (16–25%). Some TP53 PVs may facilitate HER2-derived oncogenesis in breast cancer. However, further studies with larger sample sizes are warranted to clarify the oncogenic mechanisms underlying each subtype of breast cancer in TP53 PV carriers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
TP53 and Li-Fraumeni syndrome
Tumor protein P53 (TP53) expression is induced by various physiological stresses, such as DNA damage, radiation, hypoxia, and oncogene signaling. TP53 functions as a guardian of the genome by inducing apoptosis and preventing the accumulation of genomic mutations in cells [1, 2]. Somatic pathogenic variants in TP53 are found in 18–50% of all malignant tumors in humans [3, 4], suggesting that TP53 is an early oncogenic driver in various types of cancers.
The p53 protein functions as a tumor suppressor primarily by binding to p53 DNA-binding sites in its target genes to regulate their expression [5]. TP53 pathogenic variants (PVs), both germline and somatic, are primarily distributed in the DNA-binding domain and impair TP53 transcription [6].
Li-Fraumeni syndrome (LFS) is a hereditary genetic condition attributed to TP53 PVs. It is associated with high risks for a diverse spectrum of malignant tumors, including breast cancer. The frequency of TP53 germline pathogenic variants in the general population is 0.03–0.27% [7,8,9].
Role of TP53 in breast cancer development
Breast cancer is the most frequent type of cancer observed in patients with LFS, accounting for 79% of cancers among female TP53 PV carriers [10].
In a study of human high-grade ductal carcinoma in situ (DCIS), a precursor lesion of invasive ductal carcinoma (IDC), the p53 pathway was inactivated in all DCIS specimens [11]. Breast cancer is assumed to originate from mammary epithelial cells [12]. Upon the depletion of mutant TP53 in breast cancer cells, the irregular morphology, which is a hallmark of cancer, returns to a normal mammary epithelium-like structure. This implies that mutant TP53 contributes to the disruption of the mammary tissue architecture during breast tumorigenesis [13].
Based on immunohistochemical (IHC) analyses of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), breast cancer can be classified into four basic subtypes: hormone receptor (HR: ER or/and PR) + /HER2 − , HR − /HER2 + , HR + /HER2 + , and triple-negative (negative for ER, PR, and HER2), with frequencies of 68.9%, 7.5%, 10.2%, and 13.4%, respectively [14].
TP53 PV carriers have a considerably higher rate of HER2 + tumors (67–83%) than that of noncarriers (16–25%) [15, 16]. However, the mechanism underlying HER2 overexpression in LFS-associated breast cancer remains unknown. Herein, we describe a case series of LFS in our institution and review the existing literature on TP53 functions and the mechanism underlying HER2 + breast cancer development in LFS.
Materials and methods
Case series of LFS
We conducted a retrospective review of the medical records of patients with LFS diagnosed at our institution between July 2006 and August 2021. Among 11 TP53 PV carriers, we analyzed 10 patients, excluding one unaffected carrier. We assessed the pathogenicity of TP53 germline variants reported from commercial laboratories and verified with ACMG/AMP guidelines (Table 1) [17].
Clinical criteria
The classic LFS criteria were used for the diagnosis of LFS [18]. Despite carrying a germline TP53 PV, several patients did not meet these criteria. Hence, the 2015 version of Chompret criteria [10] is widely used for identifying candidates for TP53 germline genetic testing.
Results
Case series
We identified 10 TP53 PV carriers with a history of cancer from eight families at our institution (Table 2). Cases 8–10 were from the same family, whereas the others were from different families. All patients were women, and the median age at the onset of the first tumor was 35.5 years (range: 8–45 years). Only case 2 met the classic criteria, whereas 50% of patients and 63% of families met the 2015 Chompret criteria. We found 25 tumors among the 10 patients with LFS, and the median number of tumors was two. The distribution of tumors was as follows: breast (15), bones (2), stomach (1), lung (2), colorectum (2), endometrial (1), ovary (1), and pancreas (1) (Table 2).
Case 7 was diagnosed with LFS after tumor genomic profiling. She had ovarian cancer at the age of 53 years and pancreatic cancer at the age of 54 years. The patient received somatic tumor panel testing at the age of 54 years, which indicated potential germline TP53 PVs. Thereafter, genetic testing confirmed that she carried a c.743G > A (p.R248Q) variant, the same variant detected in case 6.
Breast cancer characteristics
We observed 15 breast cancers in 9 out of 10 TP53 PV carriers (Table 2). The median age at the onset of breast cancer was 34 years (range: 26–45 years). The pathological features of the 15 breast cancers were as follows: IDC (10), invasive lobular carcinoma (ILC) (1), and DCIS (4). According to IHC analyses of 11 invasive breast cancer specimens, excluding 1 tumor that was not assessed by IHC (Case 1), 40% (4 out of 10) of tumors were HR − /HER2 + , whereas the remaining 60% were the HR + /HER2- subtype. Furthermore, among all patients with breast cancer, 56% (five of nine) had bilateral breast cancer and 67% (six of nine) had a family history of breast cancer in their mothers (Table 2).
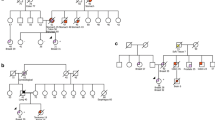
Case 8, diagnosed with ER + /PR + /HER2- breast cancer at 33 years, was a proband in this family carrying a c.473G > A (p.R158H) variant. The elder sisters of the patient (cases 9 and 10), who later underwent a TP53 analysis, also carried the same TP53 PV. In case 9, the first surveillance breast MRI detected bilateral breast cancer at the age of 43 years; DCIS in the right breast and ER + /PR + /HER2 − ILC in the left breast. Case 10 had a history of ER + /PR + /HER2 − IDC diagnosed at the age of 35 years (Table 2, Fig. 1).

Family tree of cases 8–10. According to the family history of case 8, the mother was diagnosed with breast cancer at 36 years, maternal uncle with pancreatic cancer at 60 years, and maternal grandmother with breast and gastric cancers at 45 years of age. IV-1 was an unaffected carrier with the same pathogenic TP53 variant.
Radiotherapy for breast cancer
Among 15 breast cancers, only 2 tumors were treated with radiotherapy (RT). Case 4 had bilateral breast cancer. The first tumor was left breast cancer at 26 years (Stage III, IDC, ER + /PR + /HER2 − ) treated with neoadjuvant chemotherapy with anthracycline followed by taxane, then by mastectomy with axillary lymph node dissection (Ax), with residual cancer in five nodes. Adjuvant RT and endocrine therapy were administered.
Case 6 also had bilateral breast cancer, the first tumor was left breast cancer (Stage III, IDC, ER − /PR − /HER2 +) at 28 years treated with neoadjuvant chemotherapy with anthracycline followed by a taxane plus trastuzumab regimen, and then mastectomy with Ax, with no invasive residual cancer in the breast or lymph nodes. Adjuvant trastuzumab and RT were administered.
In case 2, left breast cancer was diagnosed at 27 years and treated with breast-conserving surgery and sentinel node biopsy (no metastasis), pathological findings revealed ER − /PR − /HER2 + IDC. She did not undergo RT because we identified that she had a TP53 c.1024C > T (p.R342*) variant after surgery. A new primary breast cancer, ER + /PR + /HER2 − IDC, was detected in her remaining left breast at 38 years.
Discussion
Tumor distribution in TP53 pathogenic variant carriers
In our study, 63% of families with LFS met the 2015 Chompret criteria (Table 2) with a lower positivity rate than that previously reported; the sensitivity of the 2009 Chompret criteria is 57–82% [10, 19]. In the largest investigation of LFS in Japan (68 individuals from 48 families), 60.4% of families met the 2015 Chompret criteria [20], comparable with our results. They reported lower frequencies of soft tissue sarcoma (7.8% vs. 19.0%) and breast cancer (19.5% vs. 31.4%) in Japanese patients than in French patients with LFS [20]. In our study, 90% of patients with LFS were affected by breast cancer, accounting for 60% of all tumors (15 out of 25). Notably, these data may be biased because our institution specifically treats patients with cancer, and the number of patients with breast cancer is particularly high. This may explain why all the patients were women in this study as well as the high probability of breast cancer.
TP53 hot spots and the distribution of TP53 variants
The distribution of TP53 PVs is shown in Fig. 2. We found that 3 out of 10 patients carried variants in sites previously reported as mutation hotspots, such as R175, R245, R248, R249, R273, and R282, corresponding to the p53 DNA-binding domain [6]. Most (75%) TP53 somatic variants are missense variants [5]. In our study, 9 out of 10 patients carried a missense variant, whereas one patient carried a nonsense variant (Fig. 2).

TP53 hotspots. Six previously reported TP53 hotspots in human cancers from the IARC database are described in the lower part [6]. The spectrum of TP53 variants in our case series is shown in the upper part: green circles indicate missense variants, whereas red circles indicate nonsense variants
In our study, only case 2 had a nonsense variant; this was the only case involving a history of sarcoma and meeting the classic LFS criteria in our case series (Table 2; Fig. 2). These findings were consistent with the results obtained by Rana et al., who reported that loss-of-function variants were associated with an earlier tumor onset, increased frequency of sarcoma, and higher rate of meeting classic LFS criteria than missense variants [21].
TP53 hot spot variants facilitate HER2-derived oncogenesis
In vivo, Trp53 PV knock-in mice (R175H, R273H, and R248Q) exhibit a higher tumor bulk with an increased grade and invasion, metastatic ability, and shorter life span than those of Trp53-null mice [22, 23]. These Trp53 PVs have also been identified in humans [6] (Fig. 2).
HER2 overexpression in breast cancer activates pathways that promote cell proliferation, reduce apoptosis, and increase metastasis [24]. In vitro, p53 variants (R248Q and R273C) increase HER2 expression, whereas the suppression of PV TP53 reduces HER2 expression and inhibits the downstream pathway [25]. In a HER2 transgenic mouse model, a PV Trp53 allele induces the formation of multicentric mammary tumors and leads to early tumor onset and short survival [26].
Notably, somatic TP53 PVs are most frequent in breast cancer, reaching incidence rates of 28–37% [27, 28], and are especially frequent in the HER2 + than in the HR + subtype (72%, and 12–29%, respectively) [27]. An analysis of the HER2 + breast cancer dataset revealed that the level of HER2 mRNA expression is also considerably higher in tumors expressing TP53 PV than in tumors expressing the wild-type TP53 [29]. In our study, all HER2 + cases (4 out of 4 tumors) were graded as IHC 3 + , indicating high HER2 mRNA expression. Most HER2 IHC 3 + cases (94.7%) have been reported to show HER2 amplification by FISH [30].
These previous reports suggest that specific TP53 PVs facilitate HER2-derived oncogenesis and cancer progression in HER2 + breast cancer, potentially resulting in a high proportion of HER2 + breast cancers in patients with LFS.
Mechanisms underlying both HER2 + and HR + tumors arising in TP53 PV carriers
Although there were no HR + /HER2 + tumors in our study, HR + /HER2 + tumors are also more frequent in patients with LFS than in the general population (53% and 10%, respectively) [31, 32]. In vitro, ER binds to wild-type TP53 directly and represses its transcriptional activation [33]. Conversely, estrogen increases p53 protein levels, whereas estrogen deprivation reduces p53 levels [34]. Thus, contradictory results have been obtained regarding the relationship between ER and wild-type TP53.
Regarding the relationship between ER and TP53 PV, limited data exist; estrogen increases TP53 PV protein expression, whereas estrogen deprivation reduces TP53 expression levels [34]. In TP53 PV carriers, estrogen-ER signaling might affect the early onset of breast cancer; both ER and HER2 signaling are drivers of cell proliferation and disease progression in breast cancer, and their crosstalk might facilitate breast cancer development; however, further investigations are required to clarify these relationships.
Age at breast cancer onset in TP53 PV carriers
The median age of TP53 PV carriers at the time of diagnosis of breast cancer appears to be similar in Japan and France: the median ages were 34 years in our study, 32 years in the study by Funato et al. [20], and 33 years in a study of the French LFS working group [10]. The median age in all three independent studies was over 31 years, which is the age criterion considered for the TP53 genetic test according to the Chompret criteria.
The reported prevalence of TP53 PVs is 2.2–4.0% in women with breast cancer before the age of 31 years [35, 36]. In a study focused on HER2 + breast cancer, the prevalence of TP53 PV is 3% in patients diagnosed before 41 years; however, the prevalence increases to 8.5% in patients diagnosed before the age of 31 years [37]. In our study, the median age at the onset of breast cancer was slightly younger for HR − /HER2 + tumors than for HR + /HER2 − tumors (31 years and 35.5 years, respectively), although our study was limited by a small sample size. This suggests that TP53 genetic testing should be considered for patients with breast cancer at a slightly older age than 31 years, especially in HER2 + breast cancer. Similarly, Evans et al. suggested new criteria for TP53 germline testing to include women diagnosed with HER2 + breast cancer before the age of 36 years [38]. At our institution, TP53 germline testing is recommended for women diagnosed with breast cancer who are negative for BRCA1 and BRCA2, regardless of subtype, before 31 years of age.
Radiotherapy
TP53 PV carriers are susceptible to a high risk of radiation-induced secondary malignancy after RT. The LFS guideline recommends avoiding radiotherapy when possible [39]. In our study, two cases (cases 4 and 6) underwent adjuvant RT due to axillary lymph node metastasis and the high risk of recurrence. The patients did not have any tumors within the radiation field at follow-up times of 10 years and 9 years after RT. However, longer follow-up periods are needed because the period of radiation-induced secondary malignancy is 3–22 years (median 7 years) [40].
Mastectomy, rather than lumpectomy, is preferable to avoid a second malignancy; however, for a TP53 PV carrier with advanced breast cancer, adjuvant RT should be considered carefully depending on the risk of recurrence.
Conclusions
In summary, our study included a relatively large LFS case series from a single institution. The HER2 + breast cancer subtype was more frequent in patients with LFS (40%) than in patients with sporadic breast cancer (16–25%), consistent with previous studies. HER2 signaling is a well-known driver of cell proliferation and progression in breast cancer. Previous reports suggest that TP53 PVs facilitate HER2-derived oncogenesis, which might account for the high proportion of HER2 + breast cancer in TP53 PV carriers. It also might explain the slight difference in the onset of breast cancer between HR − /HER2 + and HR + /HER2 − tumors in our study. A limitation of our study was the small sample size at a single institution, and future investigations are warranted to clarify the oncogenic mechanisms in each subtype of breast cancer. Considering the rarity of germline TP53 PVs, clinical data collection at multiple institutions in several countries as an international collaborative study is desirable.
Data availability
Data available on request due to privacy restrictions. The variant information that support the findings of this study are openly available in Table1 and 2. Some data that support the findings of this study are available on request from the corresponding author, MH. The data are not publicly available due to restrictions their containing information that could compromise the privacy of research participants.
References
Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516–23. https://doi.org/10.1038/nature01850.
Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair. 2016;42:63–71. https://doi.org/10.1016/j.dnarep.2016.04.008.
Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. https://doi.org/10.1038/nrc2723.
Mandelker D, Donoghue M, Talukdar S, Bandlamudi C, Srinivasan P, Vivek M, et al. Germline-focussed analysis of tumour-only sequencing: recommendations from the ESMO Precision Medicine Working Group. Ann Oncol. 2019;30:1221–31. https://doi.org/10.1093/annonc/mdz136.
Yue X, Zhao Y, Xu Y, Zheng M, Feng Z, Hu W. Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J Mol Biol. 2017;429:1595–606. https://doi.org/10.1016/j.jmb.2017.03.030.
Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–86. https://doi.org/10.1101/gad.190678.112.
de Andrade KC, Mirabello L, Stewart DR, Karlins E, Koster R, Wang M, et al. Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum Mutat. 2017;38:1723–30. https://doi.org/10.1002/humu.23320.
Yamaguchi-Kabata Y, Yasuda J, Tanabe O, Suzuki Y, Kawame H, Fuse N, et al. Evaluation of reported pathogenic variants and their frequencies in a Japanese population based on a whole-genome reference panel of 2049 individuals. J Hum Genet. 2018;63:213–30. https://doi.org/10.1038/s10038-017-0347-1.
Momozawa Y, Iwasaki Y, Parsons MT, Kamatani Y, Takahashi A, Tamura C, et al. Germline pathogenic variants of 11 breast cancer genes in 7051 Japanese patients and 11,241 controls. Nat Commun. 2018;9:4083. https://doi.org/10.1038/s41467-018-06581-8.
Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33:2345–52. https://doi.org/10.1200/JCO.2014.59.5728.
Abba MC, Gong T, Lu Y, Lee J, Zhong Y, Lacunza E, et al. A molecular portrait of high-grade ductal carcinoma in situ. Cancer Res. 2015;75:3980–90. https://doi.org/10.1158/0008-5472.CAN-15-0506.
Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–46. https://doi.org/10.1046/j.1432-0436.2002.700907.x.
Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58. https://doi.org/10.1016/j.cell.2011.12.017.
Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic featues and survival. Clin Med Res. 2009;7:4–13. https://doi.org/10.3121/cmr.2009.825.
Wilson JR, Bateman AC, Hanson H, An Q, Evans G, Rahman N, et al. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J Med Genet. 2010;47:771–4. https://doi.org/10.1136/jmg.2010.078113.
Melhem-Bertrandt A, Bojadzieva J, Ready KJ, Obeid E, Liu DD, Gutierrez-Barrera AM, et al. Early onset HER2-positive breast cancer is associated with germline TP53 mutations. Cancer. 2012;118:908–13. https://doi.org/10.1002/cncr.26377.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
Li FP, Fraumeni JF Jr, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–62.
Gonzalez KD, Noltner KA, Buzin CH, Gu D, Wen-Fong CY, Nguyen VQ, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–6. https://doi.org/10.1200/JCO.2008.16.6959.
Funato M, Tsunematsu Y, Yamazaki F, Tamura C, Kumamoto T, Takagi M, et al. Characteristics of Li-Fraumeni syndrome in Japan; a review study by the special committee of JSHT. Cancer Sci. 2021;112:2821–34. https://doi.org/10.1111/cas.14919.
Rana HQ, Clifford J, Hoang L, LaDuca H, Black MH, Li S, et al. Genotype-phenotype associations among panel-based TP53+ subjects. Genet Med. 2019;21:2478–84. https://doi.org/10.1038/s41436-019-0541-y.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. https://doi.org/10.1016/j.cell.2004.11.006.
Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20:898–909. https://doi.org/10.1038/cdd.2013.17.
Sachdev JC, Jahanzeb M. Blockade of the HER family of receptors in the treatment of HER2-positive metastatic breast cancer. Clin Breast Cancer. 2012;12:19–29. https://doi.org/10.1016/j.clbc.2011.07.001.
Román-Rosales AA, García-Villa E, Herrera LA, Gariglio P, Díaz-Chávez J. Mutant p53 gain of function induces HER2 over-expression in cancer cells. BMC Cancer. 2018;18:709. https://doi.org/10.1186/s12885-018-4613-1.
Yallowitz AR, Li D, Lobko A, Mott D, Nemajerova A, Marchenko N. Mutant p53 amplifies epidermal growth factor receptor family signaling to promote mammary tumorigenesis. Mol Cancer Res. 2015;13:743–54. https://doi.org/10.1158/1541-7786.MCR-14-0360.
Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. https://doi.org/10.1038/nature11412.
Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. https://doi.org/10.1038/nature17676.
Fedorova O, Daks A, Shuvalov O, Kizenko A, Petukhov A, Gnennaya Y, et al. Attenuation of p53 mutant as an approach for treatment Her2-positive cancer. Cell Death Discov. 2020;6:100. https://doi.org/10.1038/s41420-020-00337-4.
Gown AM, Goldstein LC, Barry TS, Kussick SJ, Kandalaft PL, Kim PM, et al. High concordance between immunohistochemistry and fluorescence in situ hybridization testing for HER2 status in breast cancer requires a normalized IHC scoring system. Mod Pathol. 2008;21:1271–7. https://doi.org/10.1038/modpathol.2008.83.
Lund MJ, Butler EN, Hair BY, Ward KC, Andrews JH, Oprea-Ilies G, et al. Age/race differences in HER2 testing and in incidence rates for breast cancer triple subtypes: a population-based study and first report. Cancer. 2010;116:2549–59. https://doi.org/10.1002/cncr.25016.
Masciari S, Dillo DA, Rath M, Robson M, Weitzel JN, Balmana J, et al. Breast cancer phenotype in women with TP53 germline mutations: a Li-Fraumeni syndrome consortium effort. Breast Cancer Res Treat. 2012;133:1125–30. https://doi.org/10.1007/s10549-012-1993-9.
Sayeed A, Konduri SD, Liu W, Bansal S, Li F, Das GM. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 2007;67:7746–55. https://doi.org/10.1158/0008-5472.CAN-06-3724.
Fernandez-Cuesta L, Anaganti S, Hainaut P, Olivier M. Estrogen levels act as rheostat on p53 levels and modulate p53-dependent responses in breast cancer cell lines. Breast Cancer Res Treat. 2011;125:35–42. https://doi.org/10.1007/s10549-010-0819-x.
Mouchawar J, Korch C, Byers T, Pitts TM, Li E, McCredie MRE, et al. Population-based estimate of the contribution of TP53 mutations to subgroups of early-onset breast cancer. Australian Breast Cancer Family Study. Cancer Res. 2010;70:4795–800. https://doi.org/10.1158/0008-5472.CAN-09-0851.
Bakhuizen JJ, Hogervorst FB, Velthuizen ME, Ruijs MW, van Engelen K, van Os TA, et al. TP53 germline mutation testing in early-onset breast cancer: findings from a nationwide cohort. Fam Cancer. 2019;18:273–80. https://doi.org/10.1007/s10689-018-00118-0.
Eccles DM, Li N, Handwerker R, Maishman T, Copson ER, Durcan LT, et al. Genetic testing in a cohort of young patients with HER2-amplified breast cancer. Ann Oncol Off J Eur Soc Med Oncol. 2016;27:467–73. https://doi.org/10.1093/annonc/mdv592.
Evans DG, Woodward ER, Bajalica-Lagercratz S, Oliveira C, Frebbourg T. Germline TP53 testing in breast cancers: why, when and how? Cancers. 2020;12:E3762. https://doi.org/10.3390/cancers12123762.
Frebourg T, Bajalica Lagercrantz S, Oliveira C, Magenheim R, Gareth Evans D, European Reference Network GENTURIS. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet EJHG. 2020;28:1379–86. https://doi.org/10.1038/s41431-020-0638-4.
Hisada M, Garber TE, Fung CY, Fraumeni JF, Li FP. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90:606–11. https://doi.org/10.1093/jnci/90.8.606.
Acknowledgements
We thank the patients who participated in this study and their families.
Funding
This study received no external funding.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.H. and A.U.; data curation: K.K., H.S., and A. H.; tumor board: I.F., N.H., and S.T.; writing—original draft preparation: M.H.; writing—review and editing: T.K., H.I, T.M., Y.H., A.N., E.N., T.N., T. T., and T.U.; and supervision: S. O. and A.U. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Dr. Takayuki Ueno and Dr. Toshimi Takano are editorial board members; all other authors have no conflicts of interest.
Ethical approval
This retrospective study was approved by the Institutional Review Board of the Cancer Institute Hospital of the Japanese Foundation for Cancer Research (2022-GB-024), approved on August 4, 2022.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Hosonaga, M., Habano, E., Arakawa, H. et al. Case series of Li-Fraumeni syndrome: carcinogenic mechanisms in breast cancer with TP53 pathogenic variant carriers. Breast Cancer (2024). https://doi.org/10.1007/s12282-024-01612-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12282-024-01612-3