
Abstract
Background
Linkeropathies refers to a series of extremely rare hereditary connective tissue diseases affected by various glycosyltransferases in the biosynthesis of proteoglycans. We report for the first time two heterozygous variants of B3GAT3 in a Chinese infant, in whom Marfan syndrome was suspected at birth.
Case presentation
A 2-month-old boy from a non-consanguineous Chinese family without a family history presented severe phenotypes of joint dislocation, obvious flexion contractures of the elbow, arachnodactyly with slightly adducted thumbs, cranial dysplasia, foot abnormalities and aortic root dilation; Marfan syndrome was suspected at birth. Our patient was the youngest, at the age of 2 months, to experience aortic root dilation. Two B3GAT3 variants, NM_012200.2, c.752T>C, p.V251A and c.47C>A, p.S16*, with heterozygosity were identified in the patient by whole-exome sequencing; the variants were inherited from his parents. During close follow-up, significant changes in the cranial profile and obvious external hydrocephalus were present at the age of 7 months, which differs from previously reported cases.
Conclusion
We diagnosed a patient with congenital heart defects at an early age with a B3GAT3-related disorder instead of Marfan syndrome and expanded the spectrum of B3GAT3-related disorders. We also provide a literature review of reported B3GAT3 cases; for at least one of the variants, this is the first report of genotype–phenotype correlations in individuals with cardiovascular defects being related to the acceptor substrate-binding subdomain of B3GAT3.
Similar content being viewed by others

Background
Linkeropathies, namely, a series of rare multisystem hereditary connective tissue diseases affected by abnormal biosynthesis of proteoglycan (PGs), feature a diversity of clinical manifestations, such as short stature, skeletal deformity, brachycephaly, joint contracture and/or dislocation, craniofacial abnormalities and cardiac defects. PGs in the extracellular matrix consist of one or more glycosaminoglycan (GAG) chains attached to core proteins, playing key roles in the growth and differentiation of cells as well as cell–cell and cell–matrix interactions [1]. Secreted PGs are responsible for the structure of cartilage and bone and are closely related to the development of linkeropathies [2]. The biosynthesis of the common tetrasaccharide linker region GlcUAβ1-3Galβ1-3Galβ1-4Xyl between the core protein and the hydrophilic glycosaminoglycan side chain of PGs is a complex multi-step process that involves five genes encoding various glycosyltransferases, including XYLT1 (MIM: 608124), XYLT2 (MIM: 608125), B4GALT7 (MIM: 604327), B3GALT6 (MIM: 615291), and B3GAT3 (MIM: 606374). The beta1,3-glucuronyltransferase (GlcAT-I) encoded by B3GAT3 completes the last step of transfer of a glucuronic acid (GlcA) from the donor substrate uridine diphosphate-glucuronic acid (UDP-GlcUA) to the linkage region Gal-β-(1-3)-Gal-β-(1-4)-Xyl [1, 3]. Variants in these five genes have been reported in patients with variable phenotypes of linkeropathies [4,5,6].
GlcAT-I contains 335 residues, including a transmembrane domain, cytoplasmic region, proline-rich stem region and catalytic region, with complicated components of donor and acceptor substrate binding subdomains at residues 75-197 and 198-308, respectively [7, 8]. B3GAT3-related disorders (B3GAT3-RD) are autosomal recessive diseases characterized by dislocations of the elbows, hips, and knees, foot deformities, short stature, and cardiovascular defects. It has been reported that pathological variants related to the phenotypes of B3GAT3-RD are mainly located in the catalytic region, with substrate acceptor-binding subdomain variants tending to be associated with more severe phenotypes; conversely, donor-binding subdomain variants lead to milder phenotypes [9]. To date, 12 variants in the B3GAT3 gene have been described in 29 patients from 15 families with mild to severe phenotypes, but only half of them presented with heart defects (Fig. 1).

Structure of B3GAT3 with all reported variants. Variants associated with a phenotype of cardiovascular abnormalities are marked in red; our patient is underlined
Here, we report a 7-month-old boy with two heterozygous variants located in the acceptor substrate-binding subdomain and transmembrane domain of GlcAT-I from a nonconsanguineous healthy family in China, in whom Marfan syndrome (MFS, MIM: 154700) was suspected at birth. We also provide a literature review of clinical and molecular data in reported B3GAT3-RD patients displaying cardiovascular defects, which expands the phenotypic spectrum of B3GAT3-related disorders with cardiovascular defects.
Case presentation
The proband boy was born in a non-consanguineous Chinese family without a history of hereditary diseases. He was taken to the Guangdong Provincial People’s Hospital in Guangdong, China at 2 months of age for genetic counselling with a discharged diagnosis of suspected Marfan syndrome at birth due to arachnodactyly. He was the only baby of this Chinese family. Both parents were healthy, without a history of analogous occurrence. The pregnancy had gone well until the occurrence of gestational diabetes mellitus and decreased amniotic fluid in the last trimester. Although maternal blood glucose was well managed with diet, the amniotic fluid decreased over time. Figure 2 shows the clinical characteristics of this proband. Clinical follow-up occurred at the ages of 5 and 7 months separately. Radiological examination was also performed. We obtained permission to publish the pictures and clinical data.

Clinical manifestation in this proband. A Long fingers at age of 2 months. Clinical features at the age of 5 months; B foot abnormality; C joint flexion contracture in the right elbow, long fingers with slightly adducted thumbs; D left cubitus valgus
As mentioned above, the pregnant mother developed gestational diabetes mellitus in the third trimester that was well controlled by diet; a decrease in amniotic fluid was noticed at 30 plus weeks. The amniotic fluid index (AFI) was 7.5 cm at 38 weeks of gestation, whereas the AFI was reduced to 7.1 cm after 7 days. Consequently, caesarean section was induced at 39+ weeks of gestation due to oligohydramnios. The newborn baby survived and was in good condition, with a weight of 3000 g; the Apgar score was 10 points both at 1 min and 10 min. However, the child presented with long fingers and toes that were highly suspected to be caused by MFS. At the age of 2 months, he had a widened forehead, depressed nasal bridge, blue sclera, short neck, arachnodactyly and long toes. Echocardiography showed an increase in the diameter of the aortic sinus of 12.6 mm, but the arch of the aorta and the descending aorta were normal. When assessing development of the limbs at the age of 5 months, we discovered obvious flexion contractures of the right elbow, scoliosis, long fingers with slightly adducted thumbs, foot abnormality, and body length that was 66 cm, longer by two centimetres than the previous month, and a condition of bent limbs. The occipitofrontal circumference was 45 cm at the age of five and a half months. His cutaneous features showed reticular marble skin, especially over the thoracoabdominal skin and dorsum of hands, without any cutis laxa-like appearance, as occurs in others [9, 10].
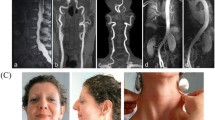
MRI was performed at the age of 7 months due to macrocephalus and a large anterior fontanel, indicating enlarged cisterns and frontotemporal sulcus. He was diagnosed by a neurosurgeon with external hydrocephalus, which could seriously affect the intellectual development and lack of available therapeutic measures. Dislocated joints of the left elbow, bilateral hips and vertebral instability of T11 and T12 were also noted by radiological examination. Symptomatic treatment and follow-up were suggested by clinicians. The clinical features and examination of this patient at the age of 7 months are shown in Fig. 3. The diameter of the aortic sinus was 16 mm, with a Z-score of 2.55 (Fig. 4).

Clinical and radiological features in the present patient at the age of 7 months. Radiological features: A enlarged head, external hydrocephalus on MRI, B Scoliosis, left elbow dislocation, radioulnar synostosis, vertebral instability on T11, and T12, and bilateral dislocation of the hip

Echocardiography showing a broadened sinus of aorta
Informed consent for whole-exome sequencing using peripheral blood from the proband and his parents was obtained. Whole-exome sequencing analysis identified two heterozygous variants in the B3GAT3 gene, c.752T>C (p.V251A) and c.47C>A (p.S16*), which was confirmed by Sanger sequencing. (Table 1, Fig. 5) The paternally inherited p.V251A variant, classified as VUS (variant of uncertain significance) according to the American College of Medical Genetics guidelines [11], has low frequencies (< 0.001) in population genomic databases and is predicted to be a “damaging” variant by SIFT (0.003) and Polyphen2-HDIV (0.725). The maternally inherited S16* variant is a likely pathogenic variant. No variants in the MFS causative genes of FBN1, ACTA2, MYLK, or MYH11 were identified, indicating that Marfan syndrome was determined to be less likely. Thus, the patient was diagnosed with B3GAT3-related disorder.

Sanger sequencing revealing c.752T>C (p.V251A) (A) and c.47C>A (p.S16*) (B)
Discussion and conclusions
Herein, we identify two B3GAT3 variants with heterozygosity in a Chinese family; this is the fifth patients being compound heterozygous reported in the literature thus far (shown in Table 3), differing from others with homozygous variant.
B3GAT3-related disorder is an extremely rare disease characterized by various manifestations varying from mild to severe. To date, only 30 patients with 14 variants in B3GAT3 have been reported in 16 families from different countries [9, 10, 12,13,14,15,16,17,18,19,20], including our patient; among cases, skeletal dysplasia and facial deformity are common, whereas cardiovascular defects only affected 14 individuals (Table 2). Among them, the most common cardiovascular abnormalities are aortic root dilation and ventricular septal defect (in 6 patients), bicuspid aortic valve (BAV; in 4), valve insufficiency, including the—mitral, tricuspid and aortic valves (—in 5), patent foramen ovale (PFO; in 5), atrial septal defect (ASD; in 3), ascending aorta dilation, patent ductus arteriosus (PDA) and pulmonary stenosis (in 1). Our patient was the youngest to display aortic-related events, namely, at the age of 2 months.
Baasanjav et al. for the first time reported Larsen-like syndrome with variable malformations of the heart with B3GAT3 variants, including BAV, aortic root dilation, mitral valve prolapse, ASD and PFO, and demonstrated expression of B3GAT3 RNA in the heart, aorta, bone and osteoblasts of mouse tissues and the presence of GlcAT-I protein in the human aorta [12]. Correspondingly, almost half of individuals with B3GAT3 variants have been reported to present heart defects, half of whom developed aortic diseases such as aortic root dilation and ascending aorta dilation (shown in Table 2). Very recently, Colman et al. [9] concluded that the two subdomains of the catalytic region of GlcAT-I are related to the phenotypes, despite only limited reports. Moreover, the more severe phenotypes tended to be associated with variants in the substrate acceptor—binding subdomain, whereas donor-binding subdomain variant manifest as milder phenotype. To date, five patients being compound heterozygous have been reported, among whom at least one variant is located in the substrate acceptor—binding subdomain. (Table 3, Fig. 1) [15, 18, 20, 21]. Among individuals with compound heterozygous variants in B3GAT3, our patient was the youngest to exhibit defined heart defects, and an enlarged sinus of the aorta is severely associated with an increased risk of death and cardiovascular complications, especially in neonates.
Based on Pedersen’s research about the structure of B3GAT3 [8], we identified that among patients with cardiovascular abnormalities, at least one variant in the acceptor substrate-binding subdomain of B3GAT3 was detected, which seems to be related to a severe phenotype (shown in Fig. 5). Variant of p.G223S occurred in six families, but only half of them presented with cardiovascular defects due to phenotypic disparity. Two patients with compound heterozygous variants, including p.R225*, exhibited more severe manifestations and lethality, without any heart defects [20]. Further insight into the relationship between altered GlcAT-I and heart defects is needed due to the limited data.
It is interesting to note that patients harboring the same variant display variable phenotypes. p.R277Q was detected in 6 patients of 2 unrelated consanguineous Arab families [12, 13]. In contrast to the five siblings in the first large family, a 5-year-old boy developed distinct manifestations of dental abnormalities, refractive error, skin wrinkling, developmental delay, and bilateral inguinal hernia. Similarly, the same p.G223S variant with extremely severe phenotypes occurred in 8 subjects of 6 families from different countries, most of whom died before the age of 1 year [9, 17, 19]. The clinical manifestations are not exactly the same, except for common symptoms of radioulnar synostosis, joint contractures, and foot deformity. More data are needed to determine whether there is a “hotspot” variant in B3GAT3.
Overlapping and diverse features of various syndromes make diagnosis difficult. Six patients suspected of having Antley–Bixler syndrome with prenatal craniosynostosis were eventually diagnosed with B3GAT3-RD by genetic testing. In the present study, based on arachnodactyly and skeletal dysplasia at birth and an enlarged aortic sinus at the age of 2, the infant was suspected of having neonatal Marfan syndrome (nMFS) [22,23,24,25], which is the most severe form of MFS. MFS is a group of rare hereditary connective tissue diseases caused by the fibrillin-1 gene (FBN1 [MIM: 134797]), featuring skeletal deformity, pectus excavatum, arachnodactyly, long, narrow face, malar hypoplasia, micrognathia, retrognathia, ocular disorder and cardiovascular abnormalities including aortic root dilation, and mitral or tricuspid insufficiency [26],—whereas nMFS mainly occurs in infants and young children. The prognosis of nMFS is so poor that it has extremely high mortality before the age of 2 years [27] and the most patients die from rapid progression of dilation of the aorta. Exons 24–32 are known as the neonatal region of FBN1, leading to a relative genotype–phenotype correlation in nMFS [28, 29] For our case, after performing whole-exome sequencing, we think it less likely to be diagnosed as MFS due to the lack of FBN1 variant. In addition, Loeys–Dietz syndrome (LDS4 [MIM: 614816], LDS5 [MIM: 615582]), Ehlers–Danlos syndrome (EDS [MIM: 130000]), and Beals syndrome (MIM: 121050) may share some specific phenotypic features of skeletal deformity and cardiovascular abnormalities [30, 31]. Therefore, identifying the cause of hereditary diseases as early as possible and making correct differential diagnoses by gene sequencing are conducive to clinical care and treatment.
The birth prevalence of congenital anomalies is estimated to be 20.8 per 1000 registered births [32];—congenital heart diseases (CHDs) are the most common, with figures varying from 2.5 to 17 per 1000 live births [33], and closely related to perinatal and infant mortality. Complex conditions comprise only ~ 15% of congenital heart diseases with a clearly known cause, while syndromic CHD with single gene variants accounts for 3–5% of congenital heart defects, such as Holt-Oram syndrome(MIM: 142900) and Noonan syndrome (NS4 [MIM: 610733]) [34]. In addition, Larsen-like syndrome caused by B3GAT3 variant involves congenital heart defects as well as skeletal dysplasia, increasing the risk of death. Maternal diabetes mellitus is one of the main causes of CHD, in which the accumulation of metabolic fuels affects the most fundamental process of cardiac development through altered gene expression [35]. In fact, we found that two pregnant mothers (including our presentation) developed gestational diabetes mellitus that was well controlled by diet during pregnancy; both sons presented variable cardiovascular defects with compound heterozygosity in B3GAT3 [15].
In conclusion, we report a 7-month-old boy with cardiovascular defects at an early age who carried two heterozygous variants in B3GAT3 and in whom MFS was suspected. The findings contribute to the spectrum of congenital cardiovascular abnormalities in B3GAT3-RD, although heart defects are not present in every case.
Availability of data and materials
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2021), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number HRA001274 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
Abbreviations
- PG:
-
Proteoglycan
- GAG:
-
Glycosaminoglycan
- B3GAT3:
-
Beta-1,3-glucuronyltransferase
- MFS:
-
Marfan syndrome
- AFI:
-
Amniotic fluid index
- nMFS:
-
Neonatal Marfan syndrome
- LDS:
-
Loeys–Dietz syndrome
- EDS:
-
Ehlers–Danlos syndrome
- CHD:
-
Congenital heart diseases
- NS:
-
Noonan syndrome
- BAV:
-
Bicuspid aortic valve
- PFO:
-
Patent foramen ovale
- ASD:
-
Atrial septal defect
- PDA:
-
Patent ductus arteriosus
References
Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537.
Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–52.
Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–51.
Bui C, Huber C, Tuysuz B, Alanay Y, Bole-Feysot C, Leroy JG, et al. XYLT1 variants in Desbuquois dysplasia type 2. Am J Hum Genet. 2014;94:405–14.
Umair M, Eckstein G, Rudolph G, Strom T, Graf E, Hendig D, et al. Homozygous XYLT2 variants as a cause of spondyloocular syndrome. Clin Genet. 2018;93:913–8.
Nakajima M, Mizumoto S, Miyake N, Kogawa R, Iida A, Ito H, et al. Variants in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am J Hum Genet. 2013;92:927–34.
Pedersen LC, Tsuchida K, Kitagawa H, Sugahara K, Darden TA, Negishi M. Heparan/chondroitin sulfate biosynthesis. Structure and mechanism of human glucuronyltransferase I. J Biol Chem. 2000;275:34580–5.
Pedersen LC, Darden TA, Negishi M. Crystal structure of beta 1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J Biol Chem. 2002;277:21869–73.
Colman M, Van Damme T, Steichen-Gersdorf E, Laccone F, Nampoothiri S, Syx D, et al. The clinical and variantal spectrum of B3GAT3 linkeropathy: two case reports and literature review. Orphanet J Rare Dis. 2019;14:138.
Alazami AM, Al-Qattan SM, Faqeih E, Alhashem A, Alshammari M, Alzahrani F, et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum Genet. 2016;135:525–40.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Baasanjav S, Al-Gazali L, Hashiguchi T, Mizumoto S, Fischer B, Horn D, et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89:15–27.
von Oettingen JE, Tan WH, Dauber A. Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy-report of the second family with B3GAT3 variant and expansion of the phenotype. Am J Med Genet A. 2014;164a:1580–6.
Budde BS, Mizumoto S, Kogawa R, Becker C, Altmuller J, Thiele H, et al. Skeletal dysplasia in a consanguineous clan from the island of Nias/Indonesia is caused by a novel variant in B3GAT3. Hum Genet. 2015;134:691–704.
Job F, Mizumoto S, Smith L, Couser N, Brazil A, Saal H, et al. Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: expanding the disease phenotype. BMC Med Genet. 2016;17:86.
Bloor S, Giri D, Didi M, Senniappan S. Novel splicing variant in B3GAT3 associated with short stature, GH deficiency, hypoglycaemia, developmental delay, and multiple congenital anomalies. Case Rep Genet. 2017;2017:3941483.
Yauy K, Tran Mau-Them F, Willems M, Coubes C, Blanchet P, Herlin C, et al. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique variant. Genet Med. 2018;20:269–74.
Ritelli M, Cinquina V, Giacopuzzi E, Venturini M, Chiarelli N, Colombi M. Further defining the phenotypic spectrum of B3GAT3 variants and literature review on linkeropathy syndromes. Genes. 2019;10:631.
Jones KL, Schwarze U, Adam MP, Byers PH, Mefford HC. A homozygous B3GAT3 variant causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am J Med Genet A. 2015;167a:2691–6.
Byrne AB, Mizumoto S, Arts P, Yap P, Feng J, Schreiber AW, et al. Pseudodiastrophic dysplasia expands the known phenotypic spectrum of defects in proteoglycan biosynthesis. J Med Genet. 2020;57:454–60.
Yap P, Liebelt JE, Amor DJ, Moore L, Savarirayan R. Pseudodiastrophic dysplasia: two cases delineating and expanding the pre and postnatal phenotype. Am J Med Genet A. 2016;170a:1363–6.
Peng Q, Deng Y, Yang Y, Liu H. A novel fibrillin-1 gene missense variant associated with neonatal Marfan syndrome: a case report and review of the variant spectrum. BMC Pediatr. 2016;16:60.
Solé-Ribalta A, Rodríguez-Fanjul X, Carretero-Bellon JM, Pascual-Sala C, Martorell-Sampol L, Bobillo-Pérez S, et al. Neonatal Marfan syndrome: a rare, severe, and life-threatening genetic disease. J Pediatr. 2019;211:221-221.e2.
Morse RP, Rockenmacher S, Pyeritz RE, Sanders SP, Bieber FR, Lin A, et al. Diagnosis and management of infantile Marfan syndrome. Pediatrics. 1990;86:888–95.
Tognato E, Perona A, Aronica A, Bertola A, Cimminelli L, De Vecchi S, et al. Neonatal Marfan Syndrome. Am J Perinatol. 2019;36:S74–6.
Dietz HC, Cutting CR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense variant in the fibrillin gene. Nature. 1991;352:337–9.
Strigl S, Quagebeur JM, Gersony WM. Quadrivalvar replacement in infantile Marfan syndrome. Pediatr Cardiol. 2007;28:403–5.
Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltone L. Variants in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nat Genet. 1994;6:64–9.
Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of variant type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 variants: an international study. Am J Hum Genet. 2007;81:454–66.
Tunçbilek E, Alanay Y. Congenital contractural arachnodactyly (Beals syndrome). Orphanet J Rare Dis. 2006;1:20.
Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L, Loeys BL. Differences in manifestations of Marfan syndrome, Ehlers–Danlos syndrome, and Loeys–Dietz syndrome. Ann Cardiothorac Surg. 2017;6:582–94.
Tennant PW, Pearce MS, Bythell M, Rankin J. 20-year survival of children born with congenital anomalies: a population-based study. Lancet. 2010;375:649–56.
van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. 2011;8:50–60.
Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. 2017;120:908–22.
Lisowski LA, Verheijen PM, Copel JA, Kleinman CS, Wassink S, Visser GH, et al. Congenital heart disease in pregnancies complicated by maternal diabetes mellitus. An international clinical collaboration, literature review, and meta-analysis. Herz. 2010;35:19–26.
Acknowledgements
The study was supported by the Department of Cardiac Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China. Thanks for the participation and support of this family in the study.
Funding
This study was supported by grants from the National Key Research and Development Program of China (2017YFC1308003) and Guangzhou Science and Technology Program (2014y2-00052). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. RF financed the research; provided guidance and supervision for the study; completed the revision of the manuscript. All the authors have nothing to disclose.
Author information
Authors and Affiliations
Contributions
Y.L. conceived and drafted the manuscript. C.Z. coordinated the study and completed the collection of clinical data. Y.L., C.Z. completed the analysis of data and the follow up of patients. H.Z., W.F., Q.W. participated in the partial revision of manuscript. R.F. provided guidance and supervision for the study. Y.L., C.Z., R.F. contributed to the discuss of results and final revision of manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present study was approved by the Research Ethics Committee, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, strictly abided by Declaration of Helsinki, national regulations in China. Written informed consent to participate was obtained from the proband’s parents.
Consent for publication
Written informed consent was obtained from the patient’s parents for the publication of this case report.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Zhang, C., Zhang, H. et al. Severe phenotypes of B3GAT3-related disorder caused by two heterozygous variants: a case report and literature review. BMC Med Genomics 15, 27 (2022). https://doi.org/10.1186/s12920-022-01160-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01160-9