
Abstract
Background
Adenylate kinase (AK) deficiency is a rare red cell enzymopathy associated with moderate to severe congenital nonspherocytic hemolytic anemia, along with mental and psychomotor retardation (in exceptional cases). Only ten mutations have been detected in the AK1 gene to date. In this study, we aimed to diagnose the unexplained issue of haemolytic anaemia and offer antenatal screening to the family.
Methods
Genomic DNA was isolated from whole blood by a standard protocol. Targeted next-generation sequencing (t-NGS) was performed to identify pathogenic variants in the patient and control samples. A chronic villus sample was collected at 11 weeks of gestation from the mother, and molecular testing was performed. Genetic confirmation was concluded by Sanger DNA sequencing. Bioinformatics tools predicted the pathogenicity of the variant.
Results
t-NGS revealed a homozygous variant (c.301C > A, p. Gln101Lys) in the AK1 gene in the patient and heterozygosity in the fetus and parental samples. The prediction tools SIFT, Polyphen2, Provean, PMUT, Mutation taster, and Mutation Assessor, confirmed the damaging effect of the variant on the AK1 protein structure
Conclusion
We have presented a novel mutation in the AK1 gene (p. Gln101Lys) associated with adenylate kinase deficiency. It is the first prenatal diagnosis of AK deficiency in India, where heterogeneity is exceptionally high.
Similar content being viewed by others

Background
Adenylate kinase (AK) deficiency (OMIM 103000) is an autosomal recessive disorder associated with moderate to severe congenital hemolytic anemia, with psychomotor impairment observed in few cases [1]. The enzyme involved in this disorder is adenylate kinase type I (AK1), which catalyzes the conversion of adenine nucleotides in the presence of Mg2+ or Mn2+: Mg2++ ATP + AMP = Mg2++ ADP + ADP.
AK1 belongs to the cytosolic enzyme family (EC 2.7.4.3). The AK1 gene is located on chromosome 9 on location 9q34. 11 (NCBI Gene ID: 203) and is highly expressed mainly in tissues with a high turnover rate, such as blood, brain, and muscles[2]. According to the available literature, certain mutations in this gene resulting in a functionally inadequate enzyme; so far, only ten mutations have been reported in the AK1 gene. [1, 3,4,5,6,7,8,9] (details are mentioned in Table 1).
This study investigated the molecular basis of erythrocyte AK deficiency in an Indian family and provided prenatal diagnosis to them for subsequent pregnancy.
Methods
Clinical history
A 5-year-old Indian boy presented with severe neonatal jaundice and severe anemia requiring regular blood transfusions was referred to us for a complete hemolytic anemia workup. He had a hepatosplenomegaly with a liver 4 cm in size and spleen up to the umbilicus. The direct and indirect Coombs tests were negative and increased serum lactate dehydrogenase level ( 3400 U/L; reference range 140–280 U/L). The haemoglobin concentration was in the range of 5–7 gm/dl (male reference range 13–16 gm/dl). He has no history of fever and skin rashes. HPLC of the patient and parents indicated the absence of haemoglobinopathies. The Peripheral blood smear suggested dimorphic anemia with predominantly saw hypochromic, normocytic cells at the initial investigation. Bone marrow examination showed erythroid hyperplasia with megaloblasts. The biochemical test for the RBC membrane protein defect, i.e. Hereditary spherocytosis, was performed using eosin 5' maleimide by flow cytometry was within the normal range (980 MCF; reference range 900–1200 MCF). The activity of erythrocyte glucose-6-phosphate dehydrogenase, pyruvate kinase, and glucose phosphate isomerase was normal (details are given in Table 2). Presently, no developmental delay or mental retardation has been observed in the patient.
Molecular studies
We collected peripheral blood from healthy controls, patients, and parents after dually signed informed consent. DNA was extracted using a standard protocol, and a targeted next-generation sequencing (t-NGS) library was generated. We performed library preparation using Illumina's TruSeq Custom Amplicon v1.5 kit (FC 130 1001) using 250 ng genomic DNA, following the manufacturer's instructions. Samples were pooled and loaded at 20 pM on MiSeq using a v3 600 cycle reagent kit sequencing 2 × 301 paired‐end reads (Illumina, San Diego, CA, USA). The library was sequenced to mean > 80-100X coverage on the Illumina MiSeq sequencing platform. The gene panel includes red cell haemoglobinopathies, enzymopathies, membrane disorders, congenital dyserythropoietic anaemias, and bone marrow failure syndrome-related genes. Numbers of genes included in the panels with corresponding accession numbers are obtained from the Single Nucleotide Polymorphism database (dbSNP at www.nchi.nlm.nih.gov/SNP), and the Ensemble Genome Browser (www.ensembl.org) are listed in Additional file 1: Table S1). The sequences obtained were aligned to the human reference genome (GRCh37/hg19) using the BWA program and analyzed using Picard and GATK version 3.6. The clinically relevant variants were annotated with the published literature and databases such as ClinVar, OMIM, GWAS, HGMD, and SwissVar. When sequence changes were found, independent PCR products were sequenced to confirm the mutations. In support that these sequence changes were not polymorphic variations, we verified that none was reported in the 1000 Genomes, https://www.internationalgenome.org/ and Human Gene Mutation Database http://www.hgmd.cf.ac.uk/ac/index.php
Prenatal diagnosis and detection of the familial AK deficiency causative mutation
A gynecologist conducted chronic Villus Sampling (CVS) during the 11th week of pregnancy of the mother. Genomic DNA was isolated using the standard protocol. We used the exon-specific primers mentioned by Dongerdiye 2020 et al. [9] for DNA amplification and Sanger sequencing of mother, father, and fetus samples.
Bioinformatics analysis
The effect of the variant was studied by multiple algorithms, such as MutationTaster, https://www.mutationtaster.org Polyphen-2, https://genetics.bwh.harvard.edu/pph2 SIFT, https://sift.jcvi.org. Mutation Assessor, https://mutationassessor.org M-CAP, http://bejerano.stanford.edu/mcap/, Combined Annotation Dependent Depletion (CADD), https://cadd.gs.washington.edu/. The probability of the mutation affects protein function was evaluated. Therefore the output “low” indicates a neutral variant. For Condel, the score ranges from 0 (neutral) to 1 (damaging). The crystallographic model of recombinant human adenylate kinase (EC 2.7.4.3) was downloaded from the Protein Data Bank (www.rcsb.org/pdb/; PDB-ID: IZ83) [6]. The impact of substitution on the structure and function of the protein was studied using PyMol software (DeLano Scientific, San Carlos, CA, USA) (http://www.pymol.org/) and Swiss Protein databank viewer (https://spdbv.vital-it.ch/).
Results
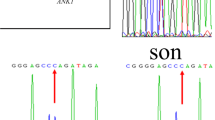
The complete details of the biochemical and hematological investigation of the patient and family are summarized in Table 2. Genetic analysis performed by the t-NGS panel revealed a single nucleotide substitution in exon 5 (c.301C > A) of AK1 gene, which caused glutamine to lysine (CAA to AAA) substitution at codon 101 (p. Gln101Lys). We observed a homozygous mutation in the proband. Parents were analyzed for the c.301C > A mutation by DNA Sanger sequencing; both parents were heterozygous for the mutation (Fig. 1). The novel variant c.301C > A, p.Gln101Lys, was submitted to the ClinVar database and submitted raw data (Accession No.: PRJNA745516: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA745516.) We measured AK enzyme activity in 50 healthy controls to the established normal range (reference range 297–360 IU/gHb), the proband (38.0 IU/g Hb), and parents' sample (mother 192.0 IU/gHb, father 208.0 IU/gHb). Biochemical findings correlated with molecular results.

Pedigree and electropherogram of the patient and family carrying the AK1 gene c.301C > A mutation
We counseled the family for the consequences of severe enzyme deficiency. Therefore, at the time of the second pregnancy, they decided to undergo prenatal screening. The fetal DNA was screened for the complete AK1 gene. DNA Sanger sequencing identified substitution c.301C > A at codon 101, causing a heterozygous change from glutamine to lysine. The pregnancy continued, and the normal healthy child was born after nine months and followed up for one year. There were no symptoms of anemia and jaundice.
The AK1 protein (PDB ID-1Z83) consists of three chains, A, B, and C, spanning 194 residues. Each chain consists of one large central "CORE" domain and two small peripheral domains, the NMP binding and the LID domains. Upon ATP binding, the LID domain closes over the phosphoryl transfer site. The amino acid residue position Q101 is an important AMP binding site and 39,44,138,149 residues. Any changes at these AMP binding sites possibly hamper the catalytic cycle of the enzyme. Figure 2a shows a complete ribbon representation of the protein (PDB ID-1Z83) with chains A, B, and C of the AK1 enzyme, along with an insight into the Q101 position helical structure (Fig. 2b) and amino acid change from wild type (glutamine) to mutant type (lysine) (Fig. 2c, d). Multiple sequence alignment confirmed that the amino acid glutamine at 101 positions is conserved across species (Fig. 2e). Most bioinformatic prediction tools demonstrate the harmful effect of the amino acid change from glutamine to lysine (Table 3 summarizes the prediction results).

a Complete ribbon representation of adenylate kinase protein (PDB ID-1Z83) with chains A, B, and C. b Secondary structure of the protein (PDB ID-1Z83) showing the amino acid residue at position 101 (Q101). c Wild type amino acid residue glutamine 101 (Q101). d Mutant type amino acid residue Lysine101 (K101). e The residue (Q) at position 101 of AK-1 is highly conserved across species
Discussion
In the present study, an Indian patient was diagnosed with rare red cell adenylate kinase deficiency. A novel homozygous (p. Gln101Lys) mutation in the AK1 gene was detected using a disease-targeted NGS panel. Previously, we reported the first case of AK deficiency from India, caused by compound heterozygous (c.71A > G and c.413G > A) mutations in the AK1 gene [9]. Clinically, these patients have incidents of chronic hemolytic anemia but no evidence of mental retardation or psychomotor impairment. Similar to other erythrocyte enzyme deficiencies, the choice of treatment depends upon the severity of the disease. Splenectomy may be recommended for severe transfusion-dependent AK deficient patients [14]. During the study, we excluded all the possible causes of hemolytic anemia, including RBC membrane defects. A recent review on red cell membrane defects mentioned that the flow‐cytometric osmotic fragility test is a new gold standard method for diagnosing HS, HE, and DHS in combination with eosin‐5'‐maleimide testing [13].
In India, only a few research groups have incorporated NGS-based genetic analysis for the routine diagnosis of RBC enzymopathy [9, 10]. However, custom NGS panels or whole-genome sequencing are widely used in various other laboratories to diagnose hemolytic anemia. The use of next-generation sequencing allows the identification of new causative genes, and polygenic conditions, and genetic factors that modify the disease severity of hereditary anemias. Disease-targeted gene panels often have higher diagnostic rates than those of exome sequencing or genome sequencing and are designed to maximize coverage, sensitivity, and specificity for the included genes [11]. The only drawback of using a custom NGS panel is that it involves a limited number of genes. Therefore, a continuous update is required for the best results. We have achieved an approximately 80% diagnostic yield using our custom panel [12]. However, these results may vary depending upon the number of patients, their clinical history, and phenotype-genotype correlations.
We identified three missense substitutions in the AK1 gene, Gln24Arg, Gln101Lys, and Arg138His, in two unrelated Indian families. These mutations could lead to dysfunction of the enzyme molecule by hampering AMP binding capacity. This study focuses on providing prenatal diagnosis to the family and gives accurate genetic advice. The fetal DNA was heterozygous for the substitution c.301C > A; p.Gln101Lys and advised to continue the pregnancy. Next-generation sequencing has many advantages, as it is cost-effective and gives high yield and speed. In contrast, there are certain limitations of NGS that significantly impact the accuracy of the results. The t-NGS panel has proven precise for our study, but its application may vary from lab to lab.
Conclusion
In conclusion, the targeted NGS panel identified a novel causative mutation in the AK1 gene in a 5-year-old male child with severe transfusion-dependent haemolytic anaemia. Identification of the pathogenic mutation helped us to offer a prenatal diagnosis in this family. This study also re-emphasizes the importance of NGS in diagnosing unexplained haemolytic anaemia in severe patients.
Availability of data and materials
All the data generated or analyzed in this study are included in this manuscript. All the gene sequences were retrieved from Ensemble Genome Browser (www.ensembl.org). We received Accession numbers for novel mutation in AK1 gene in exon 5 (c.301C > A, p. Gln101Lys) submitted on ClinVar submission Portal (Accession No: VCV001162194.2 and Variation ID: 1162194). Reference datasets used in this study is human reference genome (GRCh37/hg19), (https://www.ncbi.nlm.nih.gov/clinvar/variation/1162194/). The datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive Database (Accession No.: PRJNA745516: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA745516. The web links of the relevant datasets were as follows: hg19 http://genome.ucsc.edu/, 1000 Genomes project (http://www.1000genomes.org/), dbSNP (http://www.ncbi.nlm.nih.gov/snp), gnomAD (https://gnomad.broadinstitute.org/about), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and OMIM (http://omim.org).
Abbreviations
- ADP:
-
Adenosine diphosphate
- AK:
-
Adenylate kinase
- AMP:
-
Adenosine monophosphate
- ATP:
-
Adenosine triphosphate
- BWA:
-
Burrows-wheeler alignment
- CVS:
-
Chronic villus sample
- DNA:
-
Deoxyribonucleic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- EMA:
-
Eosin 5 maleimide
- G6PD:
-
Glucose-6-phosphate-dehydrogenase
- GATK:
-
Genomeanalysistoolkit
- GPI:
-
Glucose phosphate isomerase
- GWA:
-
Genome wide association study
- HB:
-
Haemoglobin
- HCT:
-
Hematocrit
- HGMD:
-
Human genome mutation database
- HPLC:
-
HIGH: performance liquid chromatography
- LDH:
-
Lactate dehydrogenase
- MCF:
-
Mean channel fluorescence
- MCH:
-
Mean corpuscular haemoglobin
- MCHC:
-
Mean corpuscular haemoglobin concentration
- MCV:
-
Mean corpuscular volume
- NGS:
-
Next-generation sequencing
- NMP:
-
Nucleoside monophosphate
- OMIM:
-
Online mendelian inheritance in man
- PDB:
-
Protein data bank
- PDB:
-
Protein databank
- PK:
-
Pyruvate kinase
- PLT:
-
Platelet
- PROVEAN:
-
Protein variation effect analyzer
- RBC:
-
Red blood cell
- RDW:
-
Red cell distribution width
- SIFT:
-
Sorting intolerant from tolerant
- t-NGS:
-
Targeted-next-generation sequencing
- WBC:
-
White blood cell
References
Corrons J-LV, et al. Red cell adenylate kinase deficiency: a molecular study of 3 new mutations (118G>A, 190G>A, and GAC deletion) associated with hereditary nonspherocytic hemolytic anemia. Blood. 2003;102:353–6.
Noma T. Dynamics of nucleotide metabolism as a supporter of life phenomena. J Med Investig. 2005;52:127–36.
Bianchi P, et al. A case of complete adenylate kinase deficiency due to a nonsense mutation in the AK-1 gene (Arg 107> Stop, CGA>TGA) associated with chronic hemolytic anemia. Br J Haematol. 1999;105:75–9.
Fermo E, et al. A new variant of adenylate kinase (delG138) associated with severe hemolytic anemia. Blood Cells Mol Dis. 2004;33:146–9.
Miwa S, et al. Red cell adenylate kinase deficiency associated with hereditary nonspherocytic hemolytic anemia: clinical and biochemical studies. Am J Hematol. 1983;14:325–33.
Matsuura S, et al. Human adenylate kinase deficiency associated with hemolytic anemia. A single base substitution affecting the solubility and catalytic activity of the cytosolic adenylate kinase. J Biol Chem. 1989;264:10148–55.
Qualtieri A, et al. Severe erythrocyte adenylate kinase deficiency due to homozygous A → G substitution at codon 164 of human AK1 gene associated with chronic haemolytic anaemia. Br J Hematol. 1997;99:770–6.
Niizuma H, et al. Splenectomy resolves hemolytic anemia caused by adenylate kinase deficiency. Pediatr Int. 2017;59:228–30.
Dongerdiye R, et al. Red cell adenylate kinase deficiency in India: identification of two novel missense mutations (c.71A>G and c.413G>A). J Clin Pathol. 2019;72:393–8.
Jamwal M, et al. Next-generation sequencing-based diagnosis of unexplained inherited hemolytic anemias reveals wide genetic and phenotypic heterogeneity. J Mol Diagn. 2020;22:579–90.
Russo R, et al. Genetics and genomics approaches for diagnosis and research into hereditary anemias. Front Physiol. 2020;22(11): 613559. https://doi.org/10.3389/fphys.2020.613559.
Kedar PS, et al. Study of pathophysiology and molecular characterization of congenital anemia in India using targeted next-generation sequencing approach. Int J Hematol. 2019;110:618–26.
Iolascon A, et al. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haematol. 2019;187:13–24.
Iolascon A, et al. Recommendations for splenectomy in hereditary hemolytic anemias. Haematologica. 2017;102:1304–13.
Acknowledgements
We would like to thank patients and family members for their cooperation and participation in this study. This study was performed with the support of the Indian Council of Medical Research New Delhi and the Department of Biotechnology (DBT) New Delhi for financial support. The authors' sincere thanks are due to Medgenome Laboratories Ltd. for NGS of the AK1 gene for the index case.
Funding
This study was performed with the financial support of the Indian Council of Medical Research New Delhi and the Department of Biotechnology New Delhi (DBT Project No. BT/PR20782/MED/12/737/2016). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
PK and RD1* designed the experiment. RD1*, RD2*, and PW collected samples and conducted the study. AS clinically evaluated and performed CVS for prenatal diagnosis. She also helped with genetic counseling and manuscript correction. PK and RD1* analyzed the data. PK and RD1* drafted the work and substantively revised it. *Please Note-RD1: Rashmi Dongerdiye and RD2: Rati Devendra. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee approved this study of the Institutional Ethical Committee of ICMR-National Institute of Immunohaematology, Mumbai. All procedures performed in studies involving human participants were performed per the ethical standards of the institutional review board of the Institute and followed the 1964 Helsinki declaration. We obtained written informed consent from all individual members included in the study. In a minor, we received written consent from the parents or legal guardians.
Consent for publication
We obtained written informed consent to identify images or other personal or clinical details from all the participants. In the case of a minor, we obtained written informed consent for publications identifying pictures or other personal or clinical information from the parents or legal guardians.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Gene List for Targated NGS Panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dongerdiye, R., Sampagar, A., Devendra, R. et al. Rare hereditary nonspherocytic hemolytic anemia caused by a novel homozygous mutation, c.301C > A, (Q101K), in the AK1 gene in an Indian family. BMC Med Genomics 14, 191 (2021). https://doi.org/10.1186/s12920-021-01038-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01038-2