
Abstract
Background
To strengthen the understanding of Hereditary Spherocytosis (HS) and determine the disease-causing mutation present with neonatal jaundice. HS is a hemolytic condition resulting from various erythrocyte membrane defects. Many different mutations result in HS, including mutations in ANK1.
Case presentation
A term neonate presented at ten hours with severe jaundice requiring exchange transfusion. At two months he was hospitalized due to repeated pallor and anemia requiring blood transfusions. Using next-generation sequencing, we discovered the responsible mutation in the proband but not in his parents; a heterozygous nucleotide variation of c.1000delA (p.1334Sfs*6) in ANK1. Thus hereditary spherocytosis was diagnosed.
Conclusions
Genetic detection is an important means of discovering the cause of hemolytic anemia in neonates and infants where routine diagnostic tests are unrevealing. We found a novel de novo mutation, c.1000delA (p.1334Sfs*6) in ANK1 that might account for other cases of HS in the Chinese population.
Similar content being viewed by others


Background
Hereditary spherocytosis (HS) is a relatively common inherited disorder characterized by spherical-shaped red blood cells (RBCs) [1]. The erythrocyte membrane protein defects causing HS reduce erythrocyte stability and deformability, thereby increasing fragility producing hemolysis [2]. Approximately 75% of HS cases are autosomal dominant; the remaining 25% are either autosomal recessive or de novo mutations. Its prevalence is 1 in 2,000 in people of Northern European descent [3]. The clinical manifestations of HS in the neonatal age are typically hyperbilirubinemia followed by anemia. Owing to the commonness of neonatal jaundice, the diagnosis of HS can be missed in young infants, among patients whose parents are unaffected [4]. This particularly so when spherocytic erythrocytes are not prominent on the blood smear [5, 6]. Undiagnosed HS is one of the leading causes of kernicterus. Thus, early diagnosis may decrease long-term complications.
Mutations in five different genes are recognized to cause HS. These include Ankyrin (ANK1), α-spectrin (SPTA1), β-spectrin (SPTB), band 3 (SLC4A1), and protein 4.2 (EPB42). Worldwide, ANK1 mutations are the most frequent causes of HS [7].
We diagnosed HS in an infant who had severe neonatal jaundice, where the cause was not identified until anemia had required two transfusions. We discovered a novel de novo ANK1 mutation, c.1000delA (p.1334Sfs*6). This mutation could help expand the mutational genetic diagnosis of HS in jaundiced Chinese infants.
Case presentation
Methods
Next-generation sequencing
The GenCap custom enrichment kit with exon-tiling biotinylated capture probes (My Genostics China) is used to design a blood system panel for 816 genes (Additional file 1: Table S1) including ANK1, SPTA, SPTB, EPB41 and SLC4A1.
Peripheral blood samples were obtained from the neonate and his parents, and genomic DNA was purified using the QIAamp DNA Blood Mini Kit based on the manufacturer’s recommendations (Qiagen, Germany). A minimum of 3 µg genomic DNA were subjected to Illumina DNA library preparation according to manufacturer’s protocol (My Genostics, China). The enriched libraries (fragment distribution: 350–450 bp) were pooled and sequenced using Next Seq 500 (Illumina) to obtain 2 × 150 base paired-end reads.
Raw image files were processed using Bcl2Fastq software (2.18.0.12) to generate raw reads. Raw reads were trimmed for base call quality (quality score > 20) and adapter sequences using fastq-mcf tool.
Short reads were mapped to the human hg19 reference genome assembly using the Burrows-Wheeler aligner [6] (BWA 0.7.10).Aligned reads from BAM files were visualized using the Integrative Genomics Viewer (IGV 2.3) from the Broad Institute. Picard was used to remove duplicate reads and, Genome Analysis Tool Kit [8] (GATK 2.3.4) was used to obtain single nucleotide variations (SNVs) and short indels.
Filtered candidate variants listed in TSVC format files were annotated and interpreted by ANNOVAR [9], which contains an integrated functional annotation database including 1000 Genomes project, dbSNP and Clin Var were predicted by the PolyPhen-2 and SIFT tools were used to predict the functional effects of candidate variants on proteins [10]. Synonymous mutations were removed. Common variants with a minor allele frequency (MAF) > 5% in 1000 genomes Project, ExAc or ESP6500 were removed. The variants identified using this procedure were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines [11].
Variant confirmation using Sanger sequencing
The pathogenic and likely pathogenic variants were confirmed by Sanger DNA sequencing. Sanger validation primers for ANK1 variants were designed by Primer Premier. PCR samples were visualized on agarose gels and purified. Sequences were sequenced on an ABI PRISM 3730 genetic analyzer (ThermoFisher, USA) and compared with the human reference genome assembly (hg19).
Case report and results
This male was born at 37 weeks 5 days gestation, weighing 2800 g. Apgar scores were 10 at 1 and 5 min. At ten hours he was transferred from the well baby unit to the neonatal department because of severe jaundice. His hemoglobin level was 138 g/L, reticulocyte count 4.3%, total bilirubin level 233.6 μmol/L and unconjugated bilirubin 196.7 μmol/L. Phototherapy was begun and an exchange transfusion performed in the first 24 h of life. Additional clinical information is provided in Table 1. The other routine laboratory tests were found to be negative.
Spherocytes were not identified on the blood smear, but polychromatic erythrocytes and fragmented red cells were observed. The baby’s blood type was O (+). His mother’s blood type was A (+). Coombs test and G6PD screening were negative. Nine days later he was discharged home. About 30 days later, he was noted to be anemic again and a second blood transfusion was administered. He was admitted to our department at 60 days with pallor, anemia, splenomegaly (1 cm below the left costal margin) weighting 5.9 kg.
Laboratory studies at admission included: Hb 69 g/L, mean cell volume 80.2 fL, mean corpuscular hemoglobin concentration of 354.0 g/L, and reticulocyte count 4.6%. The peripheral blood smear did not show prominent spherocytes. There is less than 1% or none erythrocytes in each microscopic field which the process of making smear also can cause (Fig. 1). The presence of microcytic erythrocytes > 7.8% can be used for HS diagnosis [12].

The peripheral blood smear
Osmotic fragility test was abnormal, consistent with a diagnosis of HS. HbF was 5.2% by alkali-resistant Hb determination. The proband’s mother, father, and extended family members were well with no history of hyperbilirubinemia or anemia.
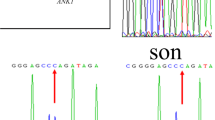
We found a novel de novo deletion ANK1 heterozygous mutation (c.1000delA, NM_000037.3) in the patient but in neither of his parents (Fig. 2). This mutation is not reported in the dbSNP database or literature. The ANK1 protein has 1881 amino acids and is composed of three structural domains: one membrane binding domain containing 24 Ankyrin repeats, one highly conserved spectrin-binding domain and a less conserved C-terminal regulatory domain. The ZU5A, ZU5B and UPA domains were identified in spectrin-binding domain and a “death” domain is found in the C-terminal regulatory domain [11].

A novel de novo deletion mutation in the ANK1gene (NM_000037.3, c.1000delA) in the patient
This frame shift mutation (c.1000delA) is located in the membrane binding domain (Fig. 3), occurring at codon 334 codon and results in a premature termination codon (PTC) at the 340 codon (p.I334Sfs*6).This PTC will create a truncated protein without spectrin-binding domain and regulatory domain and result in loss function of ANK1 gene. Moreover according to the rule of Nonsense-mediated mRNA Decay (NMD), the mutated transcript with PTC may possibly be degraded by NMD pathway.

This frame shift mutation (c.1000delA) is located in the membrane binding domain of ANK1, occurring at 334 codon (p.I334Sfs*6)
After being diagnosed with HS, the patient has received erythrocyte transfusions every month or two, with diminishing frequency. He is developing normally with no observed complications.
Discussion and conclusions
HS is a congenital hemolytic disorder due to specific erythrocyte membrane protein abnormalities [13]. In the USA Kernicterus Registry, HS was the third most common genetic cause of hemolytic anemia-related kernicterus, behind G6PD deficiency and ABO hemolytic disease. However, the majority of kernicterus cases in the Registry were on the basis of “idiopathic” severe neonatal jaundice [14].
Diagnosing HS in a jaundiced neonate whose parents do not have this disorder is challenging [15,16,17]. This is illustrated in our case, where as a neonate he had significant jaundice, and then became anemic, yet he had no family history of HS and lacked abundant spherocytes on the blood film. It was not until two months had passed and two erythrocyte transfusions were needed that he was admitted with hemolytic anemia and NGS revealed the diagnosis.
The diagnostic criterion for HS is mean corpuscular hemoglobin concentration (MCHC) in guideline for the diagnosis and management of HS [2]. But its diagnostic sensitivity and specificity are not optimal [18]. Tao et al. [19] reported that mean sphered corpuscular volume (MSCV) had diagnostic value in HS. There are limiting clinical instruments that can be used to test MSCV [18]. Xu et al. [18] and Liao et al. [20] study showed that mean reticulocyte volume (MRV) is a general and specific new index for screening HS. But firstly the unification and standardization of the reference range need to be solved. Secondly there are no available in most of laboratory including our hospital. The EMA-binding test and other methods have complicated procedures and cannot be easily conducted in a typical laboratory. The new generation of ektacytometer is difficult to be applied in the clinical laboratory, mainly used in some research centers or specialized laboratories. The using of commercial intrusion NGS to detect exactly mutation gene is not expensive in China and may be easily speeded up and popular.
It is noteworthy that the ANK1 mutation we found is both novel and de novo. De novo mutations are increasingly discovered in patients with severe early-onset diseases, such as neurodevelopmental disorders [21,22,23,24,25]. Our case supports the position that a more rigorous approach is needed to find underlying causes of “idiopathic” severe neonatal jaundice. In the Chinese population and in other populations as well, the specific mutation we identified, or other undiscovered mutations, could explain cases of severe “idiopathic” neonatal jaundice.
Availability of data and materials
The DNA sequencing data generated during the current study are deposited in the NCBI Sequence Read Archive (SRA) repository under the accession number SRR13528516 (https://www.ncbi.nlm.nih.gov/sra/?term=SRR13528516). Public databases used in this study included human hg19 reference genome assembly (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz), Human RefSeq Genome Annotation(https://www.ncbi.nlm.nih.gov/refseq/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), 1000genomes(http://www.1000genomes.org/), EXAC (http://exac.broadinstitute.org/), EVS (http://evs.gs.washington.edu/EVS), dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), HGMD (http://www.biobase-international.com/product/hgmd) and OMIM (https://www.omim.org/).
Abbreviations
- ANK1:
-
Ankyrin
- EPB42:
-
Protein 4.2
- G-6PD:
-
Glucose-6 phosphate dehydrogenase
- Hb:
-
Hemoglobin
- HS:
-
Hereditary Spherocytosis
- NGS:
-
Next Generation sequencing
- NMD:
-
Nonsense-mediated mRNA Decay
- MCV:
-
Mean cell volume
- MCHC:
-
Mean Corpuscular Hemoglobin Concentration
- MRV:
-
Mean Reticulocyte Volume
- M:
-
Month
- PTC:
-
Premature termination codon
- RBCs:
-
Red blood cells
- SPTA1:
-
α-Spectrin
- SPTB:
-
β-Spectrin
- SLC4A1:
-
Band 3
References
Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–26.
Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis–2011 update. Br J Haematol. 2012;156:37–49.
Gallagher PG. Abnormalities of the erythrocyte membrane. Pediatr Clin North Am. 2013;60:1349–62.
Delhommeau F, Cynober T, Schischmanoff PO, Rohrlich P, Delaunay J, Mohandas N, et al. Natural history of hereditary spherocytosis during the first year of life. Blood. 2000;95:393–7.
Bolton-Maggs PH. Hereditary spherocytosis; new guidelines. Arch Dis Child. 2004;89:809–12.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
He BJ, Liao L, Deng ZF, Tao YF, Xu YC, Lin FQ. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol. 2018;139:60–6.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis Toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–24.
Ipsaro JJ, Mondragón A. Structural basis for spectrin recognition by ankyrin. Blood. 2010;115:4093–101.
Mullier F, Lainey E, Fenneteau O, Da Costa L, Schillinger F, Bailly N, et al. Additional erythrocytic and reticulocytic parameters helpful for diagnosis of hereditary spherocytosis: results of a multicentre study. Ann Hematol. 2011;90:759–68.
Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27:167–78.
Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107–14.
Russo R, Andolfo I, Manna F, Gambale A, Marra R, Rosato BE, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93:672–82.
Wang X, Yi B, Mu K, Shen N, Zhu Y, Hu Q, et al. Identification of a novel de novo ANK1 R1426* nonsense mutation in a Chinese family with hereditary spherocytosis by NGS. Oncotarget. 2017;8:96791–7.
Chen J, Zhou Y, Gao Y, Cao W, Sun H, Liu Y, et al. A genetic features and gene interaction study for identifying the genes that cause hereditary spherocytosis. Hematology. 2017;22:240–7.
Xu Y, Yang W, Liao L, Deng Z, Qiu Y, Chen W, et al. Mean reticulocyte volume: a specific param eter to screen for hereditary spherocytosis. Eur J Haematol. 2016;96:170–4.
Tao YF, Deng ZF, Liao L, Qiu YL, Chen WQ, Lin FQ. Comparison and evaluation of three screening tests of hereditary spherocytosis in Chinese patients. Ann Hematol. 2015;94:747–51.
Liao L, Xu Y, Wei H, Qiu Y, Chen W, Huang J, et al. Blood cell parameters for screening and diagnosis of hereditary spherocytosis. J Clin Lab Anal. 2019;33:e22844.
De Ligt J, Willemsen MH, Van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18.
Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:241.
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–9.
Posey JE, Rosenfeld JA, James RA, Bainbridge M, Niu Z, Wang X, et al. Molecular diagnostic experience of whole-exome sequencing in adult patients. Genet Med. 2016;18:678–85.
Acknowledgements
Sanming Project of Medicine in Shenzhen (SZSM201512033).
Funding
The publishing fee was suported by Sanming Project of Medicine in Shenzhen (SZSM201512033).The analysis costs was funded by Guangdong Basic and Applied Basic Research Fund (Guangdong-shenzhen Joint Fund, 2019A1515110665).
Author information
Authors and Affiliations
Contributions
LCX designed data analysis and wrote the manuscript; ZHX provided Next-Generation Sequencing assistance; CGL edited the manuscript. SXL and FQW contributed to critical revision. All of the authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of Shenzhen Children's Hospital. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Written informed consent was obtained from the participants in the study. In the case of minors, who were younger than the age of 16, written informed consent to participate had been obtained from their parents.
Consent for publication
Written informed consent for publication of clinical details identifying images or other personal was obtained from the parents of the participant under the age of 18.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Analysis of the gene list.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, L., Xing, Z., Li, C. et al. Identification of a De Novoc.1000delA ANK1 mutation associated to hereditary spherocytosis in a neonate with Coombs-negative hemolytic jaundice-case reports and review of the literature . BMC Med Genomics 14, 77 (2021). https://doi.org/10.1186/s12920-021-00912-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00912-3