
Abstract
Background
Lung cancer is a leading cause of cancer-related death worldwide and is the most commonly diagnosed cancer. Like other cancers, it is a complex and highly heterogeneous disease involving multiple signaling pathways. Identifying potential therapeutic targets is critical for the development of effective treatment strategies.
Methods
We used a systems biology approach to identify potential key regulatory factors in smoking-induced lung cancer. We first identified genes that were differentially expressed between smokers with normal lungs and those with cancerous lungs, then integrated these differentially expressed genes (DEGs) with data from a protein-protein interaction database to build a network model with functional modules for pathway analysis. We also carried out a gene set enrichment analysis of DEG lists using the Kinase Enrichment Analysis (KEA), Protein-Protein Interaction (PPI) hubs, and KEGG (Kyoto Encyclopedia of Genes and Genomes) databases.
Results
Twelve transcription factors were identified as having potential significance in lung cancer (CREB1, NUCKS1, HOXB4, MYCN, MYC, PHF8, TRIM28, WT1, CUX1, CRX, GABP, and TCF3); three of these (CRX, GABP, and TCF) have not been previously implicated in lung carcinogenesis. In addition, 11 kinases were found to be potentially related to lung cancer (MAPK1, IGF1R, RPS6KA1, ATR, MAPK14, MAPK3, MAPK4, MAPK8, PRKCZ, and INSR, and PRKAA1). However, PRKAA1 is reported here for the first time. MEPCE, CDK1, PRKCA, COPS5, GSK3B, BRCA1, EP300, and PIN1 were identified as potential hubs in lung cancer-associated signaling. In addition, we found 18 pathways that were potentially related to lung carcinogenesis, of which 12 (mitogen-activated protein kinase, gonadotropin-releasing hormone, Toll-like receptor, ErbB, and insulin signaling; purine and ether lipid metabolism; adherens junctions; regulation of autophagy; snare interactions in vesicular transport; and cell cycle) have been previously identified.
Conclusion
Our systems-based approach identified potential key molecules in lung carcinogenesis and provides a basis for investigations of tumor development as well as novel drug targets for lung cancer treatment.
Similar content being viewed by others

Background
Lung cancer is a complex and highly heterogeneous disease involving multiple signaling pathways [1]. It is the leading cause of cancer mortality in men and the second leading cause in women worldwide [2]. Small cell lung carcinoma (SCLC) and non-small cell lung carcinoma (NSCLC) are the main types of lung cancer. The latter represents 80% of lung cancer cases and can be subclassified as squamous cell carcinoma, adenocarcinoma, or large cell carcinoma [3, 4]. Smoking is a major contributor to lung cancer development, being responsible for about 90% of cases [4]. Cigarette smoke induces inflammation and causes oxidative stress and genetic and epigenetic abnormalities that alter gene expression throughout the respiratory tract [5, 6]. Differences in gene expression in large airway epithelial cells between non-smokers and smokers have been analyzed by DNA microarray to determine the effect of smoking on the transcriptome [7]. Tobacco smoke was found to cause lung cancer by inducing of IκB kinase β- and c-Jun N-terminal kinase 1-dependent inflammation [8].
Spira et al. [9] compared gene expression data from smokers with (n = 60) and without (n = 69) lung cancer. Using a weighted-voting algorithm, these authors identified an 80-biomarker probe set that distinguished these two populations with an accuracy of 83% when validated using an independent test set (n = 52). They selected the 40 most frequently upregulated and downregulated probe sets by internal cross-validation [9]. However, this method—which uses only gene expression profiles—does not provide an integrated view. To address this issue, another study established a set of 40 biomarkers with potentially important roles in lung carcinogenesis using a network-based approach that integrated microarray gene expression profiles and information on protein-protein interactions (PPIs) [10]. Network-based approaches in the study of human disease can elucidate the genes and pathways involved as well as biomarkers and potential drug targets [11]. Network reconstruction and gene-set enrichment analysis (GSEA) have been used to mine masses of complex data obtained from genomics, proteomics, phosphoproteomics, and transcriptomics studies and organize them into a coherent global framework [12].
In this study, gene expression data from smokers with lung cancer and those without lung cancer were analyzed using a systems biology approach that included network-based and enrichment analysis of differentially expressed genes (DEGs) between normal and cancerous lung to identify potential key factors contributing to lung cancer progression.
Methods
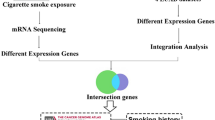
Our strategy for identifying potential key regulatory factors in smoking-induced lung cancer is shown in Fig. 1. We first identified genes that were differentially expressed between smokers with normal lungs and those with cancerous lungs. We then integrated DEG data with information obtained from a PPI database to build a network model, which we used to identify functional modules and relevant signaling pathways. Finally, we carried out a GSEA of DEGs using ChIP-x Enrichment Analysis (ChEA), Kinase Enrichment Analysis (KEA), Protein-Protein Interaction (PPI) hubs, and KEGG (Kyoto Encyclopedia of Genes and Genomes) gene-set libraries

Flow chart of systems biology approach to identify key regulatory factors in smoking-lung cancer
Dataset
Gene expression data was obtained from Gene Expression Omnibus database (DataSet Record GDS2771). Spira et al. [9] used Affymetrix HG-U133A microarrays to perform gene-expression profiling of large airway epithelial cells obtained by bronchoscopy of current and former smokers. Each individual was followed after bronchoscopy until a final diagnosis of either presence or absence of lung cancer [9]. Data included in our analysis were from smokers with lung cancer (n = 97) and those with normal lungs (n = 90).
Identifying DEGs
GEO2R and Soft parser.py analysis tools were used to identify DEGs. GEO2R uses Linear Models for Microarray Analysis R packages for background correction and normalization of gene expression data. Benjamini-Hochberg false discovery rate algorithm was used to correct for multiple testing in GEO2R [13].
Integration of DEGs with PPI database and pathway analysis using atBioNet
atBioNet identifies statistically significant functional modules using a fast network-clustering algorithm called Structural Clustering Algorithm for Networks (SCAN). atBioNet interface is connected to KEGG pathway information to allow assessment of biological functions of the modules through enrichment analysis. Each module has a pathway summary ranked according to Fisher’s exact test P value; The pathway with the lowest P value is considered as the most significant [14]. Only large DEG lists such as the combined list, GEO2R lists (top 500, 1000, and 1500 genes) were used as input lists for atBioNet, which was adjusted using the most stringent options that were not appropriate for smaller DEG lists. Of the three options for node addition, we selected the most stringent [“add only nodes directly connected to at least two input nodes (more stringent)”]. From two human PPI databases, we selected a smaller and more robust database (K2 Human Subset Database) obtained by the integration of seven original databases using K-votes approach [14].
GSEA using Enrichr
Enrichr includes 35 gene-set libraries, some of which are unique to this web server [15]. We used ChEA, KEA, PPI hubs, and KEGG gene-set libraries in this study. Enrichment was computed with the z-score method which outperformed the standard Fisher’s exact test and a combined scoring method that computed a combined P value from Fisher’s exact test and the z-score of the deviation from the expected rank [15]. As the enrichment analysis is sensitive to input genes of variable lengths, different input list sizes (from nine lists) were included to ensure that our conclusion was reliable as we concentrated on enriched items with higher overlap in these lists. Up- and downregulated gene lists, the combined list, GEO2R lists of different lengths (top 100, 250, 500, 1000, and 1500 genes) and the spira’s panel of an 80-gene biomarker [9] were used as separate input lists for Enrichr. The Spira’s panel of an 80-gene biomarker [9] was included as an independent list to enrich our study with the results of the enrichment analysis for this valuable list.
Results
Pathway analysis of DEG lists with PPI databases
We identified DEGs using GEO2R and Python script analysis tools. With GEO2R, the top DEGs were divided into different lists according to length (top 100, 250, 500, 1000, and 1500 genes). With the Python script tool, DEG lists were divided into lists of genes that were up- and downregulated as well as a list combining both of these groups. The combined list and GEO2R output lists of different lengths (500, 1000, and 1500 genes) were used as input lists for atBioNet. Top-ranked pathways for the most significant functional modules generated for each list were ranked according to the frequency percent of appearance in the top-ranked pathways lists (Fig. 2).

Top ranked pathways using atBioNet. The figure illustrates that MAPK signaling pathway is the most significant pathway related to lung cancer in smokers. Also, cell cycle, ErbB signaling pathway, glioma, insulin signaling pathway, pathways in cancer, renal cell carcinoma, and Toll-like receptor signaling pathway, and ether lipid metabolism are highly related to lung cancer
Enrichment Analysis
Up- and downregulated gene lists, the combined list, GEO2R lists of different lengths (top 100, 250, 500, 1000, and 1500 genes), and Spira’s 80-gene panel [9] were used as separate input lists for Enrichr.
Top-ranked enriched data generated for each list were ranked according to the frequency percent of their appearance in the top ten (Figs. 3, 4, 5 and 6). The transcription factors CREB1, NUCKS1, HOXB4, and MYCN frequently appeared as top-ranked transcription factors. CRX, TCF3, and GABP were predicted as novel putative transcription factors in lung cancer (Fig. 3). Enrichment analysis of kinases showed that MAPK1, IGFIR, and RPS6KA1 were the top-ranked kinases with frequency percentages of about 80% for MAPK1 and 55% for each of IGFIR and RPS6KA1. PRKAA1 was also predicted as a new putative kinase in lung cancer (Fig. 4). MAPK1, MEPCE, CDK1, MAPK3, and PRKCA frequently appeared in top 10 PPI hubs in about 70% of input lists (Fig. 5). Pathway enrichment analysis revealed MAPK signaling to be in the top ten in about 90% of input lists. Purine and ether lipid metabolism and gonadotropin-releasing hormone (GnRH) and Toll-like receptor (TLR) signaling pathways were highly related to lung cancer. Amino sugar metabolism and N-glycan biosynthesis were predicted to be dysregulated pathways in lung cancer (Fig. 6).

Transcription factors enrichment analysis using ChEA gene-set library. The transcription factors CREB1, NUCKS1, HOXB4, and MYCN frequently appeared as top-ranked transcription factors. CRX, TCF3, and GABP were predicted as novel putative transcription factors in lung cancer

Kinases enrichment analysis using KEA gene-set library. MAPK1, IGFIR, and RPS6KA1 were the top-ranked kinases with frequency percentages of about 80% for MAPK1 and 55% for each of IGFIR and RPS6KA1. PRKAA1 was also predicted as a new putative kinase in lung cancer

Enrichment analysis of PPI hubs. MAPK1, MEPCE, CDK1, MAPK3, and PRKCA frequently appeared in top 10 PPI hubs in about 70% of input lists

Enrichment analysis of pathways using KEGG gene-set library. Pathway enrichment analysis revealed MAPK signaling pathway to be in the top ten in about 90% of input lists. Purin metabolism, GnRH signaling pathway, Toll-like receptor signaling pathway, and ether lipid metabolism were highly related to lung cancer. Amino sugar metabolism and N-glycan biosynthesis were predicted to be dysregulated pathways in lung cancer
Discussion
Cancer is a complex disease and carcinogenesis in humans is a multistep process that transforms normal Cells into malignant derivatives so that investigation of the carcinogenesis from the systems perspective is inevitable [10]. Many studies have identified potential biomarkers for lung cancer using integrative approaches. Liu et al. [16] identified twelve proteins [p-CREB(Ser133), p-ERK1/2(Thr202/Tyr204), Cyclin B1, p-PDK1(Ser241), CDK4, CDK2, HSP90, CDC2p34, β-catenin, EGFR, XIAP and PCNA] which can distinguish normal and tumor samples with 97% accuracy and four proteins (CDK4, HSP90, p-CREB and CREB) which can be used to calculate the risk score of each individual patient with NSCLC to predict survival. This study identified the top six canonical pathways dysregulated in NSCLC—i.e., ATM signaling, PI3K/AKT signaling, p53 signaling, PTEN signaling, ERK/MAPK signaling, and EGF signaling. Byers et al. [17] found that SCLCs showed lower levels of several receptor tyrosine kinases and decreased activation of phosphoinositide 3-kinase (PI3K) and Ras/mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase (MEK) pathways but significantly increased levels of E2F1-regulated factors including enhancer of zeste homolog 2 (EZH2), thymidylate synthase, apoptosis mediators, and DNA repair proteins. These authors also found that PARP1 1—a DNA repair protein and E2F1 co-activator—was highly expressed at the mRNA and protein levels in SCLCs. In addition, a smoking-associated six-gene signature for predicting lung cancer risk and probability of survival has been established [4].
In this study, nine top-ranked transcription factors (CREB1, NUCKS1, HOXB4, MYCN, MYC, PHF8, TRIM28, WT1, CUX1) (Fig. 2) were found to be significant in lung cancer (Table 1) [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42], and three (CRX, GABP, and TCF3) were newly identified as potentially significant transcription factors in smoking-induced lung cancer. CRX (Cone-rod homeobox protein) has been proposed as a sensitive and specific clinical marker and potential therapeutic target in retinoblastoma and pineoblastoma [43], and is essential for growth of tumor cells with photoreceptor differentiation [44]. GABP (GA-binding protein) selectively activates the mutant TERT promoter in cancer which in turn enables cells to escape apoptosis, fundamental steps in the initiation of human cancer [45]. A TCF3-PBX1 fusion gene has been detected in adenocarcinoma in situ [46].
The top ten kinases in the present study (MAPK1, IGF1R, RPS6KA1, ATR, MAPK14, MAPK3, MAPK4, MAPK8, PRKCZ, INSR) have been previously identified (Table 2) [3, 47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63]. However, this is the first report of PRKAA1 as a significant factor in lung carcinogenesis induced by smoking. PRKAA1 (5′-AMP-activated protein kinase catalytic subunit alpha-1) mediates autophagy during differentiation of human monocytesis and can potentially serve as a therapeutic target in chronic myelomonocytic leukemia [64].
Eight proteins were significantly related to lung carcinogenesis—i.e., MEPCE, CDK1, PRKCA, COPS5, GSK3B, BRCA1, EP300, and PIN1 (Table 3) [3, 65,66,67,68,69,70,71,72,73,74,75,76]. Network analysis (Fig. 2) and enrichment analysis (Fig. 6) showed that MAPK signaling is the most significant pathway related to lung cancer in smokers. Both approaches identified that MAPK, TLR, and renal cell carcinoma signaling pathways a as being important in smoking-induced lung cancer. In addition, purine and ether lipid metabolism; GnRH, ErbB, and insulin signaling; adherens junctions; regulation of autophagy; snare interaction in vesicular transport; and cell cycle were also found to play important roles (Table 4) [77,78,79,80,81,82,83,84,85,86,87,88,89,90,91], whereas six pathways (aminosugars metabolism, dentatorubropallidoluysian atrophy, melanoma, N-glycan biosynthesis, renal cell carcinoma, and glioma) were predicted here for the first time as being significant pathways in smoking-induced lung cancer.
Increased glycolysis is a metabolic hallmark of cancer [92]. Cancer cells can reprogram glucose metabolism and hence, energy production by limiting energy metabolism to glycolysis, resulting in an aerobic glycolytic state [93]. Cancer cell metabolism is aimed at increasing biomass (e.g., nucleotides, amino acids, and lipids) to produce a new cell [94]. Melanoma, renal cell carcinoma, and glioma have all been found to be potentially related to lung cancer. Bean et al. [95] identified that targeting MET may be therapeutic target for treatment of a gefitinib/erlotinib-resistant lung tumor cell line with acquired MET amplification. Moreover, dysregulation of MET signaling has been associated with both sporadic and inherited forms of human papillary renal carcinomas [96]. The five components of the dentatorubropallidoluysian atrophy signaling pathway have been shown to have predictive power for breast cancer prognosis [97].
Conclusion
In this study, we used a systems-based approach to identify potential key molecules and pathways contributing to lung cancer progression among smokers. Three transcription factors (CRX, GABP, and TCF3) and one kinase (PRKAA1) were predicted here for the first time as being important in lung carcinogenesis. In addition, various intracellular signaling pathways and metabolic and other cellular processes were found to be closely related to lung cancer. Our findings provide new insight into the mechanisms of lung cancer development as well as potential new drug targets for disease treatment.
Abbreviations
- ChEA:
-
ChIP-x enrichment analysis
- DEG:
-
Differentially expressed genes
- KEA:
-
Kinase enrichment analysis
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein-protein interaction
References
Mani KM, Lefebvre C, Wang K, Lim WK, Basso K, Dalla-Favera R, et al. A systems biology approach to prediction of oncogenes and molecular perturbation targets in B- cell lymphomas. Mol Syst Biol. 2008;7:1–9.
Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomark Prev. 2010;19(8):1893–907.
Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18:1059–68.
Guo NL, Wan YW. Pathway-based identification of a smoking associated 6-gene signature predictive of lung cancer risk and survival. Artif Intell Med. 2012;55(2):97–105.
Van Dyck E, Nazarov PV, Muller A, Nicot N, Bosseler M, Pierson S, et al. Bronchial airway gene expression in smokers with lung or head and neck cancer. Cancer Med. 2014;3(2):322–36.
Gomperts BN, Spira A, Massion PP, Walser TC, Wistuba II, Minna JD, et al. Evolving concepts in lung carcinogenesis. Semin Respir Crit Care Med. 2011;32:32–43.
Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–8.
Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1- dependent inflammation. Cancer Cell. 2010;17:89–97.
Spira A, Beane JE, Shah V, Steiling K, Liu G, Schembri F, et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect. Lung Cancer Nat Med. 2007;13(3):361–6.
Wang YC, Chen BS. A network-based biomarker approach for molecular investigation and diagnosis of lung cancer. BMC Med Genet. 2011;4:2.
Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;7(1):56–68.
Lachmann A, Ma'ayan A. Lists2Networks: integrated analysis of gene/protein lists. BMC Bioinformatics. 2010;11:87.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300.
Ding YJ, Chen MJ, Liu ZC, et al. atBioNet—an integrated network analysis tool for genomics and biomarker discovery. BMC Genomics. 2012;13:article 325.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128.
Liu W, Wu Y, Wang L, Gao L, Wang Y, Liu X, et al. Protein signature for non-small cell lung cancer prognosis. Am J Cancer Res. 2014;4(3):256–69.
Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Dis. 2012;2:798–811.
Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Targeting CREB for cancer therapy: friend or foe. Curr Cancer Drug Targets. 2010;7:384–91.
Sakamoto KM, Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res. 2009;15(8):2583–7.
Yan Z, Shah PK, Amin SB, Samur MK, Huang N, Wang X, et al. Integrative analysis of gene and miRNA expression profiles with transcription factor-miRNA feed-forward loops identifies regulators in human cancers. Nucleic Acids Res. 2012;40:e135.
Ryu BJ, Lee H, Kim SH, Heo JN, Choi SW, Yeon JT, et al. PF-3758309, p21-activated kinase 4 inhibitor, suppresses migration and invasion of A549 human lung cancer cells via regulation of CREB, NF-kappaB, and beta-catenin signalings. Mol Cell Biochem. 2014;389:69–77.
Sargent LM, Ensell MX, Ostvold AC, Baldwin KT, Kashon ML, Lowry DT, et al. Chromosomal changes in high- and low-invasive mouse lung adenocarcinoma cell strains derived from early passage mouse lung adenocarcinoma cell strains. Toxicol Appl Pharmacol. 2008;233:81–91.
Qiu B, Han W, Tergaonkar V. NUCKS: a potential biomarker in cancer and metabolic disease. Clin Sci (Lond). 2015;128(10):715–21.
Lin SH, Wang J, Saintigny P, et al. Genes suppressed by DNA methylation in non-small cell lung cancer reveal the epigenetics of epithelial-mesenchymal transition. BMC Genomics. 2014;15:1079.
Pradhan MP, Desai A, Palakal MJ. Systems biology approach to stage-wise characterization of epigenetic genes in lung adenocarcinoma. BMC Syst Biol. 2013;7:141.
Pistoia V, Morandi F, Pezzolo A, Raffaghello L, Prigione I. MYCN: from oncoprotein to tumor-associated antigen. Front Oncol. 2012;2:174.
Wong AJ, Ruppert JM, Eggleston J, Hamilton SR, Baylin SB, Vogelstein B. Gene amplification of c-myc and N-myc in small cell carcinoma of the lung. Science. 1986;233:461–4.
Diolaiti D, McFerrin L, Carroll PA, Eisenman RN. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim Biophys Acta. 2015;1849(5):484–500.
Li K, Li Z, Zhao N, Xu Y, Liu Y, Zhou Y, et al. Functional analysis of microRNA and transcription factor synergistic regulatory network based on identifying regulatory motifs in non-small cell lung cancer. BMC Syst Biol. 2013;7:122.
Shen Y, Pan X, Zhao H. The histone demethylase PHF8 is an oncogenic protein in human non-small cell lung cancer. Biochem Biophys Res Commun. 2014;451:119–25.
Kampranis SC, Tsichlis PN. Histone demethylases and cancer. Adv Cancer Res. 2009;102:103–69.
Chen L, Munoz-Antonia T, Cress WD. Trim28 contributes to EMT via regulation of E-cadherin and N-cadherin in lung cancer cell lines. PLoS One. 2014;9(7):e101040.
Liu L, Zhao E, Li C, et al. TRIM28, a new molecular marker predicting metastasis and survival in early stage non-small cell lung cancer. Cancer Epidemiol. 2013;37:71–8.
Lin LF, Li CF, Wang WJ, Yang WM, Wang DD, et al. Loss of ZBRK1 contributes to the increase of KAP1 and promotes KAP1-mediated metastasis and invasion in cervical cancer. PLoS One. 2013;8:e73033.
Qi X-w, Zhang F, Wu H, Liu J-l, Zong B-g, Xu C, et al. Wilms’ tumor 1 (WT1) expression and prognosis in solid cancer patients: a systematic review and meta-analysis. Sci Rep. 2015;5:–8924.
Xu C, Wu C, Xia Y, Zhong Z, Liu X, et al. WT1 promotes cell proliferation in non-small cell lung cancer cell lines through up-regulating cyclin D1 and p-pRb in vitro and in vivo. PLoS One. 2013;8(8):e68837.
Wu C, Zhu W, Qian J, He S, Wu C, et al. WT1 promotes invasion of NSCLC via suppression of CDH1. J Thorac Oncol. 2013;8:1163–9.
Tsuboi A, Oka Y, Osaki T, Kumagai T, Tachibana I, Hayashi S, et al. WT1 peptide-based immunotherapy for patients with lung cancer: report of two cases. Microbiol Immunol. 2004;48:175–84.
Wong CC, Martincorena I, Rust AG, Rashid M, Alifrangis C, Alexandrov LB, et al. Inactivating CUX1 mutations promote tumorigenesis. Nat Genet. 2014;46:33–8.
Ramdzan ZM, Nepveu A. CUX1, a haploinsufficient tumour suppressor gene overexpressed in advanced cancers. Nat Rev Cancer. 2014;14:673–82.
Liu KC, Lin BS, Zhao M, Wang KY, Lan XP. Cutl1: a potential target for cancer therapy. Cell Signal. 2012;25(1):349–54.
Hulea L, Nepveu A. CUX1 transcription factors: from biochemical activities and cell-based assays to mouse models and human diseases. Gene. 2012;497:18–26.
Santagata S, Maire CL, Idbaih A, Geffers L, Correll M, et al. CRX is a diagnostic marker of retinal and pineal lineage tumors. PLoS One. 2009;4(11):e7932.
Garrisi VM, Strippoli S, De Summa S, Pinto R, Perrone A, Guida G, et al. Proteomic Profile and In Silico Analysis in Metastatic Melanoma with and without BRAF Mutation. PLoS One. 2014;9(12):e112025.
Bell RJ, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348:1036–9. doi:10.1126/science.aab0015.
Mo ML, Chen Z, Zhou HM, et al. Detection of E2A-PBX1 fusion transcripts in human non-small-cell lung cancer. J Exp Clin Cancer Res. 2013;32:29.
Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309–19. doi:10.1158/0008-5472.CAN-12-4721.
Yang L, Su T, Lv D, Xie F, Liu W, Cao J, et al. ERK1/2 mediates lung adenocarcinoma cell proliferation and autophagy induced by apelin-13. Acta Biochim Biophys Sin Shanghai. 2014;46(2):100–11.
Min H-Y, Yun HJ, Lee J-S, Lee H-J, Cho J, et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol Cancer. 2015;14:113.
Kim JS, Kim ES, Liu D, Lee JJ, Behrens C, Lippman SM, et al. Activation of insulin-like growth factor 1 receptor in patients with non-small cell lung cancer. Oncotarget. 2015;6(18):16746–56.
Fidler MJ, Shersher DD, Borgia JA, Bonomi P. Targeting the insulin-like growth factor receptor pathway in lung cancer: problems and pitfalls. Therapeutic Advances in Medical Oncology. 2012;4:51–60.
Bianconi F, Baldelli E, Ludovini V, Crinò L, Valigi P. Computational model of EGFR and IGF1R pathways in lung cancer: A systems biology approach for translational oncology. Biotechnol Adv. 2012;30(1):142–53.
Slattery ML, Lundgreen A, Herrick JS, Wolff RK. Genetic variation in RPS6KA1, RPS6KA2, RPS6KB1, RPS6KB2, and PDK1 and risk of colon or rectal cancer. Mutat Res. 2011;706:13–20.
Eisinger-Mathason TS, Andrade J, Lannigan DA. RSK in tumorigenesis: Connections to steroid signaling. Steroids. 2010;75(3):191–202.
Syljuasen RG, Hasvold G, Hauge S, Helland A. Targeting lung cancer through inhibition of checkpoint kinases. Front Genet. 2015;6:70.
Rodríguez-Ulloa A, Gil J, Ramos Y, Hernández-Álvarez L, Flores L, Oliva B, García D, Sánchez-Puente A, Musacchio-Lasa A, Fernández-de-Cossio J, Padrón G, González López LJ, Besada V, Guerra-Vallespí M. Proteomic Study to Survey the CIGB-552 Antitumor Effect. Biomed Res Int. 2015;2015:124082.
Sato A, Yamada N, Ogawa Y, Ikegami M. CCAAT/enhancer-binding protein-α suppresses lung tumor development in mice through the p38α MAP kinase pathway. PLoS One. 2013;8(2):e57013.
Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74(1):309–19.
Burkhard K, Smith S, Deshmukh R, MacKerell AD, Shapiro P. Development of Extracellular Signal Regulated Kinase Inhibitors. Curr Top Med Chem. 2009;9(8):678–89.
Kostenko S, Dumitriu G, Moens U. Tumour promoting and suppressing roles of the atypical map kinase signalling pathway ERK3/4-MK5. J Mol Signal. 2012;7:9.
Ebelt ND, Cantrell MA, Van Den Berg CL. c-Jun N-Terminal Kinases Mediate a Wide Range of Targets in the Metastatic Cascade. Genes Cancer. 2013;4(9–10):378–87.
Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta-and JNK1- dependent inflammation. Cancer Cell. 2010;17:89–97.
Liu Y, Wang B, Wang J, Wan W, Sun R, Zhao Y, et al. Down-regulation of PKCzeta expression inhibits chemotaxis signal transduction in human lung cancer cells. Lung Cancer. 2009;63:210–8.
Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G, et al. The PRKAA1/AMPKα1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy. 2015;11(7):1114–29.
Chang LC, Yu YL, Liu CY, Cheng YY, Chou RH, Hsieh MT, et al. The newly synthesized 2-arylnaphthyridin-4-one, CSC-3436, induces apoptosis of non-small cell lung cancer cells by inhibiting tubulin dynamics and activating CDK1. Cancer Chemother Pharmacol. 2015;75(6):1303–15.
Abera MB, Kazanietz MG. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol Pharmacol. 2015;87(5):832–41.
Osoegawa A, Yoshino I, Kometani T, Yamaguchi M, Kameyama T, Yohena T, et al. Overexpression of Jun activation domain-binding protein 1 in nonsmall cell lung cancer and its significance in p27 expression and clinical features. Cancer. 2006;107:154–61.
Hu MD, Xu JC, Fan Y, et al. Hypoxia-inducible factor 1 promoter-induced JAB1 overexpression enhances chemotherapeutic sensitivity of lung cancer cell line A549 in an anoxic environment. Asian Pac J Cancer Prev. 2012;13(5):2115–20.
Remsing Rix LL, Kuenzi BM, Luo Y, Remily-Wood E, Kinose F, Wright G, et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem Biol. 2014;9:353–8.
Li Z, Qing Y, Guan W, Li M, Peng Y, Zhang S, et al. Predictive value of APE1, BRCA1, ERCC1 and TUBB3 expression in patients with advanced non-small cell lung cancer (NSCLC) receiving first-line platinum-paclitaxel chemotherapy. Cancer Chemother Pharmacol. 2014;74:777–86.
Rosell R, Skrzypski M, Jassem E, Taron M, Bartolucci R, Sanchez JJ, et al. BRCA1: a novel prognostic factor in resected non-small-cell lung cancer. PLoS One. 2007;2:e1129.
Cao JX, Lu Y, Qi JJ, An GS, Mao ZB, Jia HT, et al. MiR-630 inhibits proliferation by targeting CDC7 kinase, but maintains the apoptotic balance by targeting multiple modulators in human lung cancer A549 cells. Cell Death Dis. 2014;5:e1426.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44(10):1104–10.
Yoon HE, Kim SA, Choi HS, Ahn MY, Yoon JH, Ahn SG. Inhibition of Plk1 and Pin1 by 5′-nitro indirubinoxime suppresses human lung cancer cells. Cancer Lett. 2011;316(1):97–104.
Tan X, Zhou F, Wan J, Hang J, Chen Z, Li B, et al. Pin1 expression contributes to lung cancer: Prognosis and carcinogenesis. Cancer Biol Ther. 2010;9(2):111–9.
Xu GG, Etzkorn FA. Pin1 as an anticancer drug target. Drug News Perspect. 2009;22(7):399–40710.
Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90.
Weber G. Enzymes of purine metabolism in cancer. Clin Biochem. 1983;16:57–63.
Lu C, Huang T, Chen W, Lu H. GnRH participates in the self-renewal of A549-derived lung cancer stem-like cells through upregulation of the JNK signaling pathway. Oncol Rep. 2015;34(1):244–50.
Ke X, Wu M, Lou J, Zhang S, Huang P, Sun R, et al. Activation of Toll-like receptors signaling in non-small cell lung cancer cell line induced by tumor-associated macrophages. Chin J Cancer Res. 2015;27(2):181–9.
Yang LS, Wu WS, Zhang F, Jiang Y, Fan Y, Fang HX, et al. Role of toll-like receptors in lung cancer. J Recept Signal Transduct Res. 2014;34(5):342–4.
Scott CC, Heckman A, Nettesheim P, Snyder F. Metabolism of ether-linked glycerolipids in cultures of normal and neoplastic rat respiratory tract epithelium. Cancer Res. 1979;39(1):207–14.
Xing C, Zhang R, Cui J, et al. Pathway crosstalk analysis of non-small cell lung cancer based on microarray gene expression profiling. Tumori Journal. 2015;101(1):111–6.
Hasima N, Ozpolat B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death and Disease. 2014;5(11) doi:10.1038/cddis.2014.467.e1509.
Lan Q, Hsiung CA, Matsuo K, et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet. 2012;44:1330–5.
Jung JJ, Inamdar SM, Tiwari A, Choudhury A. Regulation of intracellular membrane trafficking and cell dynamics by syntaxin-6. Biosci Rep. 2012;32:383–91.
Vincenzi B, Schiavon G, Silletta M, Santini D, Perrone G, Di Marino M, et al. Cell cycle alterations and lung cancer. Histol Histopathol. 2006;21(4):423–35.
Hoque MO, Brait M, Rosenbaum E, Poeta ML, Pal P, Begum S, et al. Genetic and epigenetic analysis of erbB signaling pathway genes in lung cancer. J Thorac Oncol. 2010;5:1887–93.
Ray A, Alalem M, Ray BK. Insulin signaling network in cancer. Indian J Biochem Biophys. 2014;51(6):493–8.
Singh S, Prakash YS, Linneberg A, Agrawal A. Insulin and the Lung: Connecting Asthma and Metabolic Syndrome. J Allergy (Cairo). 2013;2013:627384.
Petridou ET, Sergentanis TN, Antonopoulos CN, et al. Insulin resistance: an independent risk factor for lung cancer? Metab Clin Exp. 2011;60(8):1100–6.
Hanahan D, Weinberg RA. Hallmarks of cancer. the next generation. Cell. 2011;144:646–74.
Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–7.
Giubellino A, Linehan WM, Bottaro DP. Targeting the Met signaling pathway in renal cancer. Expert Rev Anticancer Ther. 2009;9:785–93.
Ma S, Kosorok MR. Detection of gene pathways with predictive power for breast cancer prognosis. BMC Bioinformatics. 2010;11:1.
Acknowledgements
We thank the reviewers of our manuscript for careful reading and for giving beneficial suggestions. We would like to thank Editage for English language editing.
Funding
No funding was available for our study.
Availability of data and materials
The data set is publicly available at Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/), under data source access GSE4115. GEO2R, atBioNet, and Enrichr are online tools. Soft parser.py was created by Ma’ayan Laboratory, Icahn school of medicine at Mount Sinai. Spira’s panel of an 80-gene biomarker is available at (https://drive.google.com/open?id=0B_fmfGY06nfgY2ZKdlRDdW4wS1E). Data supporting the conclusions of this article are included within the article.
Authors’ contributions
SE and ME designed the methods of this article. SE carried out the data analysis and wrote the paper. SE, ME, AM, and MH have been involved in drafting the manuscript and revising it. AH and HA were in the supervision committee. All the authors approved and agreed to the authorship of the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable because the study is a secondary analysis of existing public data.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
El-aarag, S.A., Mahmoud, A., Hashem, M.H. et al. In silico identification of potential key regulatory factors in smoking-induced lung cancer. BMC Med Genomics 10, 40 (2017). https://doi.org/10.1186/s12920-017-0284-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-017-0284-z