
Abstract
Background
Systems biologists are faced with the difficultly of analyzing results from large-scale studies that profile the activity of many genes, RNAs and proteins, applied in different experiments, under different conditions, and reported in different publications. To address this challenge it is desirable to compare the results from different related studies such as mRNA expression microarrays, genome-wide ChIP-X, RNAi screens, proteomics and phosphoproteomics experiments in a coherent global framework. In addition, linking high-content multilayered experimental results with prior biological knowledge can be useful for identifying functional themes and form novel hypotheses.
Results
We present Lists2Networks, a web-based system that allows users to upload lists of mammalian genes/proteins onto a server-based program for integrated analysis. The system includes web-based tools to manipulate lists with different set operations, to expand lists using existing mammalian networks of protein-protein interactions, co-expression correlation, or background knowledge co-annotation correlation, as well as to apply gene-list enrichment analyses against many gene-list libraries of prior biological knowledge such as pathways, gene ontology terms, kinase-substrate, microRNA-mRAN, and protein-protein interactions, metabolites, and protein domains. Such analyses can be applied to several lists at once against many prior knowledge libraries of gene-lists associated with specific annotations. The system also contains features that allow users to export networks and share lists with other users of the system.
Conclusions
Lists2Networks is a user friendly web-based software system expected to significantly ease the computational analysis process for experimental systems biologists employing high-throughput experiments at multiple layers of regulation. The system is freely available at http://www.lists2networks.org.
Similar content being viewed by others


Background
Experimental biologists who incorporate high-content profiling experiments within their research often face the difficultly of understanding results from many different experiments, under different conditions, and at different layers of regulation. Results from such experiments report the activity of groups of genes that function together to give rise to changes in cellular phenotype. It is often desired to compare the results from studies such as mRNA expression microarrays, ChIP-chip or ChIP-seq (ChIP-X), RNAi screens, proteomics and phosphoproteomics in one coherent global framework. Several advanced data mining techniques have been developed to address the challenge of analyzing the complexity of such datasets. Approaches fall into different categories which include: network reconstruction, gene-set enrichment analyses, and dynamical modeling [1]. Gene-set enrichment analyses are probably the most practical and successful approach so far. With this approach groups of genes identified experimentally are associated with prior biological knowledge to identify overlap similarity with sets of annotated genes for the purpose of suggesting functional biological themes [2, 3]. The usefulness of such analyses was demonstrated first in what is now a classical study by Mootha et al. [4] who examined changes in gene expression in muscle biopsies from diabetic patients to discover a group of genes associated with oxidative phosphorylation that are coordinately down regulated in diabetic muscles. Their study led to the development of the Gene Set Enrichment Analysis (GSEA) software and the development of the MSigDB database, a database containing over 5,000 lists of mammalian gene-sets where each gene-set is associated with a specific experiment or a specific common biological function used to label each gene-set [5].
A recent review by Huang, Sherman and Lempicki [3] enlisted 68 bioinformatics tools that perform enrichment analyses on lists of genes. Most of these tools use the Gene Ontology (GO) database [6] as the only source for gene-sets with specific functional category. However, several other enrichment tools are capable of linking gene lists with other annotated gene-sets, i.e., pathway databases such as KEGG [7], BioCarta, and GenMAPP [8]. Systems such as GSEA [5], DAVID [9], GFINDer [10], WebGestalt [11] are similar to the software Lists2Networks (L2N) we present here in a sense that they go beyond Gene Ontology enrichment analysis to integrate other types of biological prior knowledge. For example, DAVID, which started as a Gene Ontology enrichment tool, now provides enrichment analysis capabilities to examine lists of genes for enrichment in pathways, structural domains, protein interactions, disease association, tissue expression and more [9]. Another important feature that is implemented in some of these systems is the visualization of the overlap among lists. For example, BioVenn is a web-based tool that takes as input two or three lists and draws Venn diagrams showing the overlap among lists of genes [12]. Another feature that is only provided by few systems is the ability to manipulate and merge lists. Such functionality exists, for example, in WebGestalt which allows users to apply set operations such as union, complementation, and intersection on loaded lists [11].
All gene-set enrichment tools compute statistics of whether the overlap among two sets of genes is significant. This is an important feature since it is possible for two lists of genes to have some overlap but such overlap can be due to chance. Computing the statistical significance of overlap among pairs of gene-set lists can be achieved using different contingency table statistics such as Fisher exact, binomial proportions, or Chi-squared tests. These tests compute the probability of finding the same genes in two or more experiments, or in two or more functional categories. The Fisher exact test computes this statistics exactly whereas the binomial proportions or Chi-squared tests are approximations. The goal of applying these tests is to find gene-sets with unexpected significant overlap, an overlap which deviates from what is expected by chance. Regardless of the test used, the results are often similar but are sensitive to list size [13]. Alternatively, in many cases the ranking of genes/proteins identified experimentally is known. For example, the fold-change or p-value of the observed gene/protein level as it is compared with an experimental control. Hence, several enrichment tools also use non-parametric tests such as Kolmogorov-Smirnov and Wilcoxon tests to compute enrichment with consideration of ranks [5]. For the Lists2Networks system we only implemented the Fisher exact test to compute gene-list enrichment and overlap [12]. The Fisher exact, the binomial proportions, or the Chi-squared tests are based on the null normal distribution assumption which treats gene as independent. Such an assumption is obviously inaccurate but it simplifies the task and capable of producing useful results.
Implementation and Results
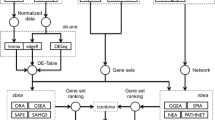
The Lists2Networks (L2N) system is implemented using a combination of PHP, JSP/Java, and JavaScript. The data is stored on the server side in a MySQL database. To make the application have a look-and-feel of a desktop application Asynchronous JavaScript and XML (AJAX) is used for concurrent updating of parts of the web-site without full page refreshes. The system contains several modules (Fig. 1) which include: an interface to upload lists, an interface to expand lists using background networks, a tool to manipulate lists using set operations, an interface to analyze lists for overlap and enrichment of functional terms, a protein-protein interaction browser, and a user communication system which allows users to share lists with other users. The following is a brief description of the different components/modules of the system:

System architecture of the L2N software. The system contains several related components (blue ellipses). Registered users are able to upload lists of genes/proteins. Set operations allow users to create new lists from existing lists. Overlap analyses use background databases of gene-list libraries to compute enrichment. Lists of genes are able to be expanded using background knowledge networks of PPIs, co-expression, and co-annotations. Users have the capability to share their lists with other users and communicate with other users through a messaging system.
The starting page and the user communication system
One of the obvious advantages of a web-based system is the ability to access data from any desired location, not being bound to a single computer. Additionally, data and analysis can be shared in a collaborative way. The start page of L2N provides users with the ability to communicate with other researchers using an integrated messaging system. For this we use a similar approach found on popular social networking sites such as Facebook. The system provides users with the ability to locate other users through a user search utility. Once users identify each-other, a friendship can be initiated by a friend request. After establishing a friendship, users can exchange messages and share gene lists. A message-board displays incoming messages and gene lists sharing requests. By accepting a gene-list, sent by another user, the list is automatically integrated into the user-lists-workspace, ready to be analyzed by the analysis components of the system.
The upload component
After starting the system for the first time, the user-lists-workspace is empty. In order to populate the workspace, gene lists have to be uploaded to the system. The upload component of the system allows users to upload lists of mammalian genes in Entrez Gene Symbol format. L2N implements four upload options within the upload component of the system: The first option allows users to drag-and-drop multiple text files containing lists of genes into a Java applet. This feature allows fast upload by bypassing the restriction of HTML forms. Alternatively, a standard HTML form can be used. Both the Java Applet and the HTML form allow for annotation of the uploaded gene-list with a detailed text-based description. The third and fourth options for uploading lists of genes are self contained and do not required user data. The third option allows users to enter any search term into a PubMed search. The system uses PubMed e-utilities to return a set of abstracts that match the searched term. These abstracts are converted to a list of human Entrez gene symbols using GeneRifs. GeneRifs is a manually curated dataset that links publications with genes. The resultant gene list can be uploaded into the workspace. The final and forth upload option uses Gene Ontology. Here users can type biological terms in a search box. The matching terms from the Gene Ontology database with the associated genes are then displayed and made ready for upload into the workspace.
The expand-lists component
This component of the L2N system provides users with the ability to expand lists based on networks created from known protein-protein interactions, co-expression correlations, or co-annotation correlations. These background knowledge networks are represented as graphs made of nodes and links [14, 15]. Interactions from those networks are used for "connecting" the genes/proteins from input lists similarly to the way we achieved this for the software system Genes2Networks [16]. The shortest paths between pairs of nodes (genes) from the input list are found to form a subnetwork that "connects" the input list nodes using additional genes/nodes from the background network (Fig. 2). The resultant subnetworks are visualized using a Flash-based interactive network viewer that is embedded within the application. Additionally, the output subnetwork, besides being visualized within the web-page, is made available for download in SIF format, amenable for import, analysis and visualization with Cytoscape [17]. Furthermore, the subnetwork that is generated from the input list is automatically converted into a new list that can be added back into the user's workspace as an expanded list. The subnetwork reconstruction process and implementation also contains features that give users the flexibility to set a threshold for inclusion of intermediate nodes and links in the subnetwork. The threshold settings are based on the specificity of the additional proteins/genes (intermediates) to interact with the input list, as well as the number of steps/links used to connect the nodes. The specificity calculation is using the proportions of links to seed nodes from the intermediate nodes compared with total interactions for the intermediate nodes in the background respective network. Intermediates are ranked based on their counts of links in the subnetwork as compared with their total links in the background prior knowledge protein-protein, co-expression or co-annotation networks.

Bottom center: Screenshot from the overlap component showing sample analysis. The background knowledge category chosen is KEA kinase-substrate enrichment. Top right: Expand Lists component screenshot. Uploaded lists can be expanded using protein-protein interactions network, co-expression network, or co-annotation network. Users can set two parameters: sensitivity and number of steps. This allows controlling the size of the expanded lists. Top left: Network of lists representation. Uploaded user lists (blue) are connected if they overlap significantly. Green and black nodes represent labeled gene-lists from a specific library (kinases and their known substrates) that overlap with the uploaded lists. Green nodes represent annotated lists that overlap with only one input list whereas black nodes are lists that overlap with two or more input lists.
The Expand Lists component gives users the ability to choose the background network to use when expanding lists. There are three options: a protein-protein interactions network, a co-expression network, and a co-annotation network. The protein-protein interaction network is compiled from a variety of experimentally determined mammalian (mouse/rat/human) interactions recorded in the following databases: BioGRID [18], Reactome, Biomolecular Interaction Network Database (BIND) [19], the Human Protein Reference Database (HPRD) [20], IntAct [21], Database of Interaction Proteins (DIP) [22], Molecular INTeractions database (MINT) [23], PDZBase [24], Protein-Protein Interaction Database (PPID) [21, 25], as well as the interactions described in references [26–28]. All interactions from these databases/datasets were determined experimentally and include a PubMed reference to the primary source article. For creating the co-expression network we used COXPRESdb [29], a database which contains a downloadable table of co-expressed genes in mouse and human. To create the co-annotation network we defined a pair-wise distance between genes:

The co-annotation is based on the co-appearance in annotated gene-set lists from MSigDB [5]. The pair-wise dependency between genes can be represented as a graph where nodes are genes and edges represent the co-appearance level between two genes in respect to inclusion in an annotated gene-set list.
The set operation component
Once lists have been uploaded, and if desired expanded, users can apply set operations on lists to generate additional lists. This feature of the system is useful for performing common steps in the analysis of many different experimental data scenarios. For example, it is often desired to obtain a consensus list of genes that appeared in a set of repeated experiments, i.e., genes that appeared to be consistently up-regulated in several microarray experiments. For applying such operation, users can apply the "intersection" function. Similarly, analysis of proteomics data often requires removal of sticky non-specific proteins, for example, removing all ribosomal proteins, which commonly reappear in immuno-precipitation followed by mass-spectrometry (IP-MS) type of experiments. For applying this operation users can apply the "not" function.
The overlap component
The overlap component of L2N is the most extensive, useful and powerful feature of the system. With this feature users can select certain lists from their library of lists to generate an overlap representation between the loaded lists, as well as overlap with categories of prior biological knowledge in the form of collections of labeled gene-lists stored in the Gene Matrix Transposed (GMT) flat file format [5]. Each GMT file contains rows of gene sets where the first two columns in the file describe the list, while the rest of the entries in each row are Entrez Gene symbols. Similarity among lists is computed using the Fisher exact test and the overlap is visualized as a distance table matrix (Fig. 2). The resultant similarity matrix displays the overlap among user-selected lists and libraries of gene-sets.
The overlap analysis section allows the identification of biological themes that can be associated with multiple lists from the user's-list-workspace. Moreover, lists from the workspace can be studied for similarity to each other and to previously annotated gene-sets from many biological categories (Gene Ontology, Pathways, microRNAs, protein domains, kinases, etc.). A complete list of prior biological knowledge gene-list libraries is provided in Table 1. To start the analysis, a GMT file which represents a category of a biological theme has to be selected. As an example, we show the results when the user chooses the kinase-substrate prior knowledge category (KEA_kinases) applied on a sample set of user inputted lists (Fig. 2). Before the analysis can begin, the user needs to select the gene lists that should be included in the overlap analysis. After selecting the lists and pressing the large green arrow, the overlap matrix is computed and displayed right next to the listings of the gene lists.
The screenshot in Fig. 2 shows the results after a sample overlap matrix was created. The matrix itself is interactive. By hovering over the squares in the matrix, information about the content of each square is displayed on the right panel. The large green numbers in Fig. 2 are the p-values of the overlap enrichment computation using the Fisher exact test before applying the Bonferroni or Benjamini-Hochberg corrections. By clicking on the red squares from the row of red squares, columns are sorted by their p-values which represent overlap with annotated gene-list libraries. After the sorting is done a table of ranked enriched terms is displayed on the right panel. The row of red squares, used for sorting, separates the matrix into two separate sections. Below the red line is the overlap between the input gene lists and the lists belonging to the chosen biological category. Each column of the matrix represents one of the input files, whereas each row below the red line represents a labeled gene list from a prior knowledge library. It is also possible to click on any of the squares of the matrix to see the genes that overlap. The matrix allows fast browsing of enriched functional annotations that match many input lists. Furthermore, the enrichment of terms from different categories associated with many input lists can be compared easily where common biological themes can be identified.
In addition to the overlap matrix display, users can view overlap between input lists as a network. Such network can be displayed by clicking the "Show Network" button on top of the overlap matrix (Fig. 2). This network visualization displays the input files as nodes, as well as enriched gene lists from a specific prior knowledge category as nodes. Only gene lists that are enriched (having high overlap) with at least one other list after the Bonferroni correction with a p-value < 0.05 are connected with an edge and included in the network for visualization. An edge (link) in the network represents a significant overlap between pairs of lists. Input lists are colored in blue, enriched gene lists from a prior knowledge category are in green or black. Black nodes are gene lists from the prior knowledge category that have significant overlap with more than one input list. In the example in Fig. 2 the biological category is KEA_kinases and it shows that different lists of proteins from the input lists are associated with different kinases.
The data for the prior biological knowledge enrichment analyses was created using original GMT files we developed, as well as few GMT files downloaded from MSigDB [5] (Table 1). The original GMT files (gene-list libraries) that we created are: pathways from WikiPathways, data from ChIP experiments, predicted microRNA-mRNA interactions from miRBase and TragetScan, kinase-substrate interactions from KEA, protein-metabolite interactions from HMDB, disease genes from OMIM, disease-gene neighborhoods using OMIM and Genes2Networks, protein interaction hubs using Genes2Networks and protein structural domains using PFAM and InterPro. Additional available libraries previously created by others are: pathways from KEGG, BioCarta and GenMAPP, as well as chromosomal location [30]. To generate the microRNAs GMT file we processed the data from miRBase [31] and TargetScan databases. Such databases contain gene lists predicted to be regulated by microRNA families. For the kinases, we used a database of experimentally determined kinase-substrate interactions we recently developed for KEA [32] by consolidating several web-based resources reporting kinase-substrate relations. The metabolites GMT file was created from data downloaded from HMDB [33], and the disease neighborhood GMT file was created from lists of genes from OMIM [34] and expanded using protein-protein interactions as described above. Expanding lists of disease genes using known protein-protein interactions assisted us in discovering SHOC2 as a novel Noonan Syndrome disease causing gene [35] justifying the disease gene neighborhood concept. The gene-lists libraries will be updated manually periodically. Specifically we are mostly interested in updating the protein-protein interactions data, kinase-substrate interactions data, datasets from RNAi and ChIP screening, and microRNA-mRNA target interactions. Such datasets will be quality controlled using manual and automated filtering methods. Users are welcome to contribute gene-list libraries to the system. However, these contributions will be monitored by the authors for quality.
The list sharing component
Since the system is web-based, we provide users with the ability to share lists and communicate results and messages with other users through a dedicated messaging system. The system provides users with the ability to locate other users through a user search utility. Once users identify each other and want to communicate and share lists with one another, a friendship request message can be initiated. Such request needs to be approved by the requested party for establishing communications. Once such friendship has been established, both users can share lists and exchange messages.
The protein-protein interactions browser component
Additional feature that is desired by experimental and computational biologists is to explore which proteins directly or indirectly interact with a specific protein of interest. It is also desired to see how lists of interactors of one protein overlap with other experimentally developed lists. For example, results from IP-MS proteomics experiments, pulling down and characterizing interactions for specific protein baits are logically compared to already known interactions for specific proteins based on literature and other resources that previously characterized protein-protein interactions. This can be used to assess how consistent the IP-MS results are with what is already known about protein-protein interactions with the bait. For this, L2N has a protein-protein interactions browser feature where users can quickly identify all direct interactors for a specific gene/protein. Users can upload lists of interactors as input lists for comparison, enrichment, expansion, and visualization, as part of the integrated analysis provided by the other parts of the L2N system. The implementation of such browser is delivered as a dynamical text-based expansion system where the original gene/protein is selected from a list and then the lists of direct interactors are dynamically displayed in a recursive manner. Protein-protein interactions have been compiled as described above.
Flash based network viewer
To visualize networks within a web-page in a dynamic representation, we used Flash/ActionScript3 which allows the efficient development of interactive web content. The advantage of using Flash over other recent web technologies such as JAVA applets and AJAX is that Flash/ActionScript3 integrates the classes of Sprites, which are a powerful vector graphics entity with attached action listeners for user interaction. Since the latest version of ActionScript (AS3), the programming language used in Flash is no longer restricted as with previous versions. AS3 has strong emphasis on visual output and user interactivity, making it ideal for dynamic web-based network visualization purposes. The network viewer is implemented using a force directed layout algorithm to place nodes by minimizing a stress function considering optimal edge length and node repulsion.
Case study: integrating proteomics and phosphoproteomics studies applied to profile embryonic stem cell differentiation
To illustrate how L2N can be utilized to integrate results from different but related high-content genome-wide profiling studies, we created a case study (Additional file 1). We integrated and analyzed data from the following four proteomics and phosphoproteomics studies applied to profile differentiating mouse and human embryonic stem cells: Lu et al. [36] who profiled the nuclear proteome after silencing of Nanog; two phosphoproteomics studies of human embryonic stem cells driven to differentiate by two different methods [37, 38]; and the Nanog interactome as determined by a serial set of proteomics experiments [39]. Although our focus and aim of the case study is to demonstrate to novice users the capabilities of the L2N software system, we obtained some interesting results. For example, there are 23 proteins that overlap between the Nanog-KO-Nuclear-Day5-Up from the Lu et al. study and the Brill et al. list of phosphoproteins identified four days after inducing differentiation with retinoic acid. This is a statistically significant overlap with a p-value of ~0.000002 (Fisher exact test). The proteins from this list are great candidates for further functional experimental validation and characterization as components of an early differentiation pathway. Additionally, to further identify proteins that potentially belong to the Nanog interactome, we cross referenced an expanded subnetwork made of the Nanog interactome reported by Wang et al. and the expand list feature of L2N with the Lu et al.-Day5-Down-List. We found that EED, JARID1B, PNO1, SMARCA5 and UTF1 are identified in both lists from such cross-reference analysis. These candidates should be further validated as bona-fide self-renewal components belonging to the Nanog interactome. EED and JARID1B are already known components of the self-renewal machinery as was discovered recently (more details can be found in the Case Study provided with this manuscript as Additional file 1).
Conclusions
Lists2Networks is a user friendly powerful system that is expected to significantly ease the computational analysis process of experimental biologists employing high-throughput experiments at multiple layers of regulation. It simplifies gene-list enrichment analyses and provides means to expand lists with three types of prior biological knowledge networks, apply list operations, browse protein-protein interactions and communicate lists between users. The ability to form networks of lists based on list similarity is a powerful method to integrate many different experiments with background knowledge for hypothesis generation. While other similar tools may already exist, L2N has several features not implemented in existing systems or partially implemented in other systems. These include, for example, the ability to expand lists using protein-protein interactions, co-expression data, and co-annotation data; the ability to share lists and communicate by a messaging system in a collaborative fashion; web-based application of list operations such as union, complementation and subtraction; never before assembled prior knowledge gene-lists libraries such as: data collected from high-throughput ChIP experiments, expanded disease gene networks, protein interaction hubs, and kinase substrates; uploaded lists are saved on the server not requiring users to re-upload lists every time they used the system whereas users can manually edit the content of lists with built-in synonym resolution feature. Additionally, users can create original gene lists using any search term through PubMed and GeneRifs or easily integrate lists from the Gene Ontology database.
In regards to future plans, we can implement the system for other model organisms and improve the ID matching features. Additionally, we plan to provide plug-ins that would enable the exporting and importing of lists using other software systems. In summary, L2N stands out being user-friendly, web-based, robust and simple, making it more accessible for novice users as compared with other existing academic or commercial packages aim to address similar needs.
Availability and Requirements
Project name: Lists2Networks
Project home page: http://www.lists2networks.org
Programming languages: PHP, Java, MySQL, ActionScript/Flash
Requirement: Web browser and internet access, Java support, JavaScript enabled browser, Flash Player 10
References
Ma'ayan A: Network integration and graph analysis in mammalian molecular systems biology. IET Systems Biology 2008, 2(5):206–221. 10.1049/iet-syb:20070075
Nam D, Kim S-Y: Gene-set approach for expression pattern analysis. Brief Bioinform 2008, 9(3):189–197. 10.1093/bib/bbn001
Huang DW, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acids Res 2009, 37(1):1–13. 10.1093/nar/gkn923
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al.: PGC-1[alpha]-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003, 34(3):267–273. 10.1038/ng1180
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al.: Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005, 102(43):15545–15550. 10.1073/pnas.0506580102
Gene Ontology Consortium: The Gene Ontology (GO) database and informatics resource. Nucl Acids Res 2004, 32(suppl_1):D258–261. 10.1093/nar/gkh036
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M: KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucl Acids Res 1999, 27(1):29–34. 10.1093/nar/27.1.29
Doniger S, Salomonis N, Dahlquist K, Vranizan K, Lawlor S, Conklin B: MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biology 2003, 4(1):R7. 10.1186/gb-2003-4-1-r7
Dennis G, Sherman B, Hosack D, Yang J, Gao W, Lane H, Lempicki R: DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biology 2003, 4(9):R60. 10.1186/gb-2003-4-9-r60
Masseroli M, Martucci D, Pinciroli F: GFINDer: Genome Function INtegrated Discoverer through dynamic annotation, statistical analysis, and mining. Nucl Acids Res 2004, 32(suppl-2):W293–300. 10.1093/nar/gkh432
Zhang B, Kirov S, Snoddy J: WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucl Acids Res 2005, 33(suppl_2):W741–748. 10.1093/nar/gki475
Hulsen T, de Vlieg J, Alkema W: BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 2008, 9(1):488. 10.1186/1471-2164-9-488
Fury W, Batliwalla F, Gregersen PK, Wentian L: Overlapping Probabilities of Top Ranking Gene Lists, Hypergeometric Distribution, and Stringency of Gene Selection Criterion. Engineering in Medicine and Biology Society, 2006 EMBS '06 28th Annual International Conference of the IEEE: 2006 2006, 5531–5534.
Ma'ayan A: Insights into the organization of biochemical regulatory networks using graph theory analyses. J Biol Chem 2008, 284(9):5451–5455. 10.1074/jbc.R800056200
Ma'ayan A, Blitzer RD, Iyengar R: TOWARD PREDICTIVE MODELS OF MAMMALIAN CELLS. Annual Review of Biophysics and Biomolecular Structure 2005, 34(1):319–349. 10.1146/annurev.biophys.34.040204.144415
Berger SI, Posner JM, Ma'ayan A: Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics 2007, 8(1):372. 10.1186/1471-2105-8-372
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Research 2003, 13(11):2498–2504. 10.1101/gr.1239303
Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M: BioGRID: a general repository for interaction datasets. Nucl Acids Res 2006, 34(suppl_1):D535–539. 10.1093/nar/gkj109
Bader GD, Betel D, Hogue CWV: BIND: the Biomolecular Interaction Network Database. Nucl Acids Res 2003, 31(1):248–250. 10.1093/nar/gkg056
Mishra GR, Suresh M, Kumaran K, Kannabiran N, Suresh S, Bala P, Shivakumar K, Anuradha N, Reddy R, Raghavan TM, et al.: Human protein reference database--2006 update. Nucl Acids Res 2006, 34(suppl_1):D411–414. 10.1093/nar/gkj141
Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, Ceol A, Moore S, Orchard S, Sarkans U, von Mering C, et al.: The HUPO PSI's Molecular Interaction format[mdash]a community standard for the representation of protein interaction data. 2004, 22(2):177–183.
Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D: DIP: the Database of Interacting Proteins. Nucl Acids Res 2000, 28(1):289–291. 10.1093/nar/28.1.289
Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, Cesareni G: MINT: a Molecular INTeraction database. FEBS Letters Protein Domains 2002, 513(1):135–140. 10.1016/S0014-5793(01)03293-8
Beuming T, Skrabanek L, Niv MY, Mukherjee P, Weinstein H: PDZBase: a protein-protein interaction database for PDZ-domains. Bioinformatics 2005, 21(6):827–828. 10.1093/bioinformatics/bti098
Grant SG: Systems biology in neuroscience: bridging genes to cognition. Current Opinion in Neurobiology 2003, 13(5):577–582. 10.1016/j.conb.2003.09.016
Rual J-F, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al.: Towards a proteome-scale map of the human protein-protein interaction network. 2005, 437(7062):1173–1178.
Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck F, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S: A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell 2005, 122(6):957–968. 10.1016/j.cell.2005.08.029
Ma'ayan A, Jenkins S, Neves S, Hasseldine A, Grace E, Dubin-Thaler B, Eungdamrong N, Weng G, Ram P, Rice J: Formation of Regulatory Patterns During Signal Propagation in a Mammalian Cellular Network. Science 2005, 309(5737):1078–1083. 10.1126/science.1108876
Obayashi T, Hayashi S, Shibaoka M, Saeki M, Ohta H, Kinoshita K: COXPRESdb: a database of coexpressed gene networks in mammals. Nucl Acids Res 2008, 36(suppl_1):D77–82.
Ogata H, Goto S, Fujibuchi W, Kanehisa M: Computation with the KEGG pathway database. Biosystems 1998, 47: 119–128. 10.1016/S0303-2647(98)00017-3
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ: miRBase: microRNA sequences, targets and gene nomenclature. Nucl Acids Res 2006, 34(suppl_1):D140–144. 10.1093/nar/gkj112
Lachmann A, Ma'ayan A: KEA: kinase enrichment analysis. Bioinformatics 2009, 25: 684–686. 10.1093/bioinformatics/btp026
Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, et al.: HMDB: a knowledgebase for the human metabolome. Nucl Acids Res 2009, 37(suppl_1):D603–610. 10.1093/nar/gkn810
Hamosh A, Scott AF, Amberger J, Bocchini C, Valle D, McKusick VA: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucl Acids Res 2002, 30(1):52–55. 10.1093/nar/30.1.52
Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, et al.: Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 2009, 41(9):1022–1026. 10.1038/ng.425
Lu R, Markowetz F, Unwin RD, Leek JT, Airoldi EM, MacArthur BD, Lachmann A, Rozov R, Ma/'ayan A, Boyer LA, et al.: Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature 2009, 462(7271):358–362. 10.1038/nature08575
Brill LM, Xiong W, Lee K-B, Ficarro SB, Crain A, Xu Y, Terskikh A, Snyder EY, Ding S: Phosphoproteomic Analysis of Human Embryonic Stem Cells. Cell Stem Cell 2009, 5(2):204–213. 10.1016/j.stem.2009.06.002
Van Hoof D, Muñoz J, Braam SR, Pinkse MWH, Linding R, Heck AJR, Mummery CL, Krijgsveld J: Phosphorylation Dynamics during Early Differentiation of Human Embryonic Stem Cells. Cell Stem Cell 2009, 5(2):214–226. 10.1016/j.stem.2009.05.021
Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH: A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444(7117):364–368. 10.1038/nature05284
Acknowledgements
This research was supported by NIH Grant No. 1P50GM071558-01A27398 and start-up fund from Mount Sinai School of Medicine to AM. We thank Amin Mazloom, Ben MacArthur, Shan Zhao and Seth Berger for help in creating GMT files.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AL and AM designed the system. AL implemented the system. AM wrote the manuscript.
Electronic supplementary material
12859_2009_3544_MOESM1_ESM.PDF
Additional file 1: Lists2Networks Case Study: Integration of Proteomics Data Applied to Embryonic Stem Cells. A demonstration of how to use Lists2Networks for the analysis of protein lists collected from several proteomics and phosphoproteomics studies that profiled differentiating mouse and human embryonic stem cells. (PDF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lachmann, A., Ma'ayan, A. Lists2Networks: Integrated analysis of gene/protein lists. BMC Bioinformatics 11, 87 (2010). https://doi.org/10.1186/1471-2105-11-87
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-11-87