
Abstract
Background
Cryptosporidium parvum (C. parvum) is a cosmopolitan parasite that infects various livestock animals including cattle. Microsatellite typing tools for identification of C. parvum subtypes are currently employed to better understand the species-specific epidemiology of cattle cryptosporidiosis. The aim of this study was to analyse the population genetics of C. parvum strains infecting cattle and recognise geographical distribution and time-span correlations in subtype prevalence in Poland. In total, 1601 faecal samples were collected from 2014 to 2018 from healthy cattle from dairy, meat and mixed breeds at the age of 1 week to 4 months. The 267 farms visited were randomly selected and represented all Polish provinces. PCR–RFLP based identification of C. parvum at the 18 small subunit ribosomal RNA (SSU rRNA) locus was performed, followed by strain subtyping by GP60-PCR.
Results
The overall prevalence of C. parvum in Polish cattle was estimated at 6.2% (100/1601). Animals below the age of 1 month were the major host for this parasite. Excluding one breed, that of dairy-meat mixed, there were no significant differences observed between breed and presence of C. parvum infections (95% TPIAll breeds: 1.67–73.53%; POPR = 0.05—0.95). Infected animals were detected in 15 out of 16 Polish provinces, with significant regional prevalence diffrences (Kruskal–Wallis rank sum test, Kruskal–Wallis χ2 = 13.46, p < 0.001). When the population genetics of C. parvum strains were analysed, 11 parasite subtypes from the IIa and IId genetic families were identified. Compared to other parasite strains, IIaA17G1R1 and IIaA17G2R1 appeared at statistically significantly higher frequency (F-test, F = 3.39; p = 0.0003). The prevalence of C. parvum subtypes in cattle was breed-related (Chi-squared test, χ2 = 143.6; p < 0.001).
Conclusions
The analysis of the population genetics of C. parvum subtypes showed that strains from the IIa subtype family predominated in the tested cattle population. However, relations in changes of subtype prevalence and circulation over time were observed. They were associated with the disappearance of some strains and emergence of new variants from the same genetic family in different geographical locations.
Highlights
C. parvum subtypes from the IIa allele family predominated in the tested cattle. The prevalence of C. parvum subtypes in cattle was breed-related. Dynamicity in the population C. parvum strains circulating in cattle was shown.
Similar content being viewed by others


Background
Among the parasitic diseases of cattle, cryptosporidiosis is of major importance [1, 2]. Symptomatic cryptosporidiosis is mainly associated with C. parvum infections [3,4,5,6]. The prevalence of C. parvum infections in cattle herds in Europe has been found to range from 1.2 to 100% and has more often been observed in diarrhoeic animals [7, 8]. Outside Europe, they have also occurred with varied prevalences ranging from 5 to 50% in North America [9, 10], 3.6 to 97.3% in South America, 0.3 to 93% in Africa [11, 12] and 3.4 to 93% in Asia [13, 14].
In Poland, the first attempt to assess the prevalence of Cryptosporidium infection in cattle was undertaken by Rzeżutka et al. in 2013 [15]. The overall prevalence of Cryptosporidium infection in the tested animal population was estimated at 17%; however, C. parvum was not the most frequently detected parasite species, being discovered in 5.1% of cases. The analysis of the microsatellite GP60 region of the C. parvum genome has revealed a various number of circulating subtypes belonging to three subtype families, which were IIa, IId, and IIl before 2014 [16]. Other investigations are in accordance regarding the circulation of the IIa subtype family, it having been the most frequently detected C. parvum subtype in European cattle [17,18,19]. Reports in the literature also note that the IId and IIl genetic families have been found less frequently [3, 4, 20, 21], and the pattern in Poland was similar as found by Kaupke and Rzeżutka [16], C. parvum strains of IIa subtype predominating compared to the sporadically appearing IId and IIl strains. In research up to the time of writing, the C. parvum IIaA17G1R1, IIaA17G2R1 and IIaA15G2R1 strains have been most often identified. Of note is that IIaA15G2R1 has also appeared as the dominant subtype in several European cattle populations [19, 20, 22,23,24,25,26,27]. C. parvum can cause infections in various hosts including humans [28]. A major role in parasite transmission is played by cattle, which are the natural reservoir of the parasite [29,30,31,32]. It has been shown that C. parvum infections in humans are more common in areas of intensive livestock farming [4].
Although numerous studies have been conducted that aimed at identification of C. parvum subtypes of bovine origin, our knowledge of population genetics of strains of this parasite and their geographical prevalence is still not complete. Furthermore, the population structure and subtype distribution in livestock as they relate to host age also require analysis. The aim of this study was to analyse the population and geographical distribution of C. parvum strains infecting cattle in Poland. Additionally, changes of subtype prevalence and circulation over time in the population of Polish cattle were investigated.
Results
Detection and age-related ocurrence of C. parvum infections in cattle
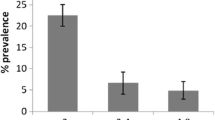
The 18 SSU rRNA gene fragment was successfully amplified in 725 out of 1601 cattle faeces samples. After NdeI digestion of all Cryptosporidium-positive samples, a restriction pattern of 18 SSU rRNA amplicons characteristic of C. parvum was shown for 100 DNA samples. Two samples contained a mixture of different parasite sequences including that of C. parvum. Their subsequent treatment with MboII confirmed identification of C. parvum and revealed the presence of C. bovis. The overall prevalence of C. parvum in Polish cattle was estimated at 6.2% (100/1601). C. parvum infections were detected in all age groups of cattle at 11.9% (63/527) frequency at 1–4 weeks, 4.9% (23/464) at > 4–8 weeks and 2.3% (14/610) at > 8–16 weeks. A relationship was observed between animal age and frequency of infection (Chi-squared test, χ2 = 1439.2, p < 0.001). Animals below the age of 1 month were the major host for this parasite and the number of C. parvum-positive cattle decreased with animal age.
Breed related prevalence of C. parvum
The sampled animals mainly represented dairy breeds (74.8%), meat and mixed dairy–meat breeds being far smaller proportions (respectively 14.5% and 10.7%). Animals of Polish Black and White Holstein Friesian (HO), Limousie (LM), mixed exclusively meat breed (MM), Simentaler (SM), mixed exclusively dairy breed (MS) and mixed dairy–meat breed (MDM) breeds were found positive for C. parvum DNA, the majority of infections (74/1601) being detected in dairy cattle of HO breed (Table 1). There were no significant differences observed between cattle breeds (95% true prevalence estimate (TPI)All breeds: 1.67–73.53%; POPR = 0.05–0.95) in appearance of C. parvum infections, with one exception: animals of MDM breed differed from the other breeds (95% TPIMDM breeds: 2.79–9.25%; POPR ≤ 0.05 and POPR ≥ 0.95). Mixed C. parvum and C. bovis infections were not associated with any particular breed (95% TPIAll breeds: < 0.0001–42.54%).
Geographical distribution of C. parvum infections
The infected animals were reared on 44 (16.5%) out of the 267 monitored farms and were located over the whole country. All evaluated farms in the Zachodniopomorskie (ZP) and Dolnośląskie (DS) provinces appeared to be C. parvum positive. None of the tested cattle from Śląskie province (SL) herds was C. parvum positive. Infections with this parasite were detected in farms located across 15 Polish provinces with varied prevalences (Table 2, Fig. 1). Significant differences in regional prevalences of C. parvum infections between the groups of provinces (high, medium and low) in Poland were shown (Kruskal–Wallis rank sum test, Kruskal–Wallis χ2 = 13.46, p < 0.001). Pomorskie (PM), ZP, Łódźkie (LD), Podkarpackie (PK) and Mazowieckie (MZ) provinces were characterised by higher prevalence of infections (16.7 ≥ 9.3%) and in descending order of prevalence the remaining provinces were Wielkopolskie (WP), Podlaskie (PL), Kujawsko-Pomorskie (KP), Lubelskie (LB), Warmińsko-Mazurskie (WM) (medium prevalence < 5.6–3.7%) and Lubuskie (LS), Opolskie (OP), Świętokrzyskie (SK), Małopolskie (MP), DS and SL (low prevalence < 2.8%). The highest frequency of parasite infections was in PM at 16.7% and the lowest in DS province at 1.1%, where only 1 out of 15 farms kept infected animals (Table 2).

The geographical distribution of C. parvum strains in Poland. Values in brackets indicate at number of detected parasite strains of particular subtypes. The percentage values shown on the map indicate at C. parvum prevalence in particular province. DS Dolnośląskie, KP Kujawsko-Pomorskie, LB Lubelskie, LD Łódzkie, LS Lubuskie, MP Małopolskie, MZ Mazowieckie, OP Opolskie, PK Podkarpackie, PL Podlaskie, PM Pomorskie, SK Świętokrzyskie, SL Śląskie, WM Warmińsko-Mazurskie, WP Wielkopolskie, ZP Zachodniopomorskie, Nd Not determined
Identification of C. parvum subtypes
The subtype was successfully identified by PCR amplification with GP60 primers for 82 out of 100 detected parasite strains but was not for the other 18 C. parvum strains. Strains from the IIa subtype family dominated with 80 affiliations and strains from the IId subtype were the other 2 successful identifications. The IIaA17G1R1 (n = 26) and IIaA17G2R1 (n = 24) strains were detected in the greatest numbers. Three subtypes, IIaA21G1R1 (n = 1), IIaA18G3R1 (n = 1) and IIaA21R1 (n = 1), were only sporadically found. The other detected strains were IIaA15G2R1 (n = 10), IIaA16G1R1b (n = 9), IIaA22G1R1 (n = 4), IIaA10G1R1 (n = 2), IIaA14G2R1 (n = 2) and IIdA24G1 (n = 2). None of the tested samples contained more than one C. parvum GP60 subtype. The ANOVA showed that only C. parvum IIaA17G2R1 and IIaA17G1R1 infections occurred at significantly higher frequency than other parasite subtypes (F-test, F = 3.39; p = 0.0003). The infections caused by IIaA17G2R1 (total number of infections 24) and IIaA17G1R1 (total number of infections 26) dominated in the studied animal population. In the case of the remaining C. parvum subtypes (total number of infections from 1 to 10, LSD0.05 = 14.1) there were no significant differences observed in the frequency of their prevalence. Mixed infections caused by two or more parasite subtypes were detected neither at the farm nor at animal level.
The nucleotide sequence analysis of the GP60 gene fragment gene showed 100% sequence identity for C. parvum strains within the following IIa subtype groups (A10G1R1, A15G2R1, A16G1R1b, A17G1R1, A17G2R1, and A24G1). C. parvum IIaA14G2R1 strains revealed 96.4% mutual nucleotide sequence identity. A higher genetic resemblance (99.6–100%) was shown for IIaA22G1R1 strains. The phylogenetic relationships between strains representing different parasite subtypes were presented on Fig. 2.

The phylogenetic maximum likelihood tree constructed using the nucleotide sequences (294 bp) of the GP60 gene fragment of C. parvum strains detected in cattle. C. hominis was used as an outgroup to root the tree. C. parvum strains detected in cattle from Poland before 2014 are marked by black diamonds
Age- and breed-related prevalence of C. parvum subtypes in cattle
There was no relationship observed between infections caused by a particular C. parvum subtype and cattle age group (Chi-squared test, χ2 = 29.4; p = 0.133). Nevertheless, particular subtypes were only shown in animals at a certain age. For instance, the IIaA15G2R1, IIaA21G1R1, IIaA10G1R1, IIaA18G3R1, IIaA21R1 and IIaA24G1 subtypes were detected in cattle below 4 weeks of age, while IIaA14G2R1 only occurred in animals between the ages of 4 and 8 weeks (Table 3). Two closely related subtypes, IIaA17G1R1 and IIaA17G2R1, caused infections in calves from all age groups. A significant relationship was shown between the detection of a specific C. parvum subtype and cattle breed (Chi-squared test, χ2 = 143.6; p < 0.001), whereby 10 out of 11 C. parvum subtypes were found in HO calves, C. parvum IIaA22G1R1 strains were only detected in LM cattle and IIaA24G1 only in MM animals (Table 3).
Geographical distribution of C. parvum subtypes
Animals infected with C. parvum were detected on 44 (16.48%) of the 267 farms. Two commonly detected IIa subtypes, i.e., IIaA17G1R1 and IIaA17G2R1, were found respectively in 8 and 6 out of the 16 Polish administrative provinces. In comparison to IId subtypes, the two aforementioned strains from the IIa allele families occurred frequently over almost the entire country except in the LS, SL and DS provinces (Fig. 1). They were present on 25 (9.36%) of the investigated farms. The IId subtypes were only found on one farm in the LS province. The relationship between the C. parvum subtype and the place of its detection was confirmed by a Pearson’s chi-squared test (χ2 = 312.0; p < 0.001). The IIaA17G1R1 strains prevailed in PK and SK, while the IIaA17G2R1 did in MP, MZ and WM. On cattle farms in ZP and LS, IIaA16G1R1b and IIdA24G1 infections comprised the majorities of observed infections, respectively.
Time-related changes in geographical prevalence of C. parvum strains
Analysing the structure of the population genetics of C. parvum subtypes in cattle in Poland, the disappearance of the strains IIaA14G1R1, IIaA18G1R1c and IIaA19G1R1 was observed with time. In their place, the new variants IIaA18G3R1 and IIaA14G2R1 representing the same genetic family appeared; however, they did so in different geographical locations. Based on the current results, it can be assumed that some subtypes from the IIa (A19G1R1) and IIl (A19R3) genetic families are not currently present in Poland. In the place of the parasite strains IIdA22G1b, IIdA23G1, and IIdA24G1c, which before 2014 had been detected in cattle herds in western Poland (ZP and PM provinces) as well as in LB province in the east of the country, a single subtype IIdA24G1 was found at the time of this investigation. Its appearance was limited to only LS in western Poland. Strains of the IId classification did not spread to other geographical locations in the country, which may indicate their endemic prevalence. For the IIa strains prevalent in Polish cattle, a change in their population dynamics related to the time and place of their detection was observed.
Before 2014, the IIaA15G2R1 subtype was circulating in livestock almost nationwide, also being the only parasite subtype detected in DS, SL, LD, and SK provinces [16]. From 2014 until the conduct of this research, infections caused by C. parvum strains of this subtype continued to affect the cattle population in Poland, although their prevalence was limited to only PL and ZP. Also in the same period of time, in the population of IIaA16 strains, two groups of variants were found, i.e., IIaA16G1R1b and IIaA16G3R1. The IIaA16G1R1b subtype prevailed and it was only present in the WP province. Currently, its spread to other regions such as ZP, PK, and PL is observed, which means transmission from the western to the eastern part of Poland. Of note is that infections in cattle caused by this subtype are endemic in the WP province. Over a period of 8 years the frequency of IIaA17G1R1 and IIaA17G2R1 infections was high and they are still considered dominant C. parvum subtypes in cattle in Poland. In many cases, they co-circulated in a given geographical area. Furthermore, the constant presence of the IIaA17G1R1 strains in WM, PM and ZP and of the IIaA17G2R1 strains in MZ, PL and WM may indicate the endemic nature of these infections.
Discussion
Although different Cryptosporidium species have been detected in cattle, symptomatic cryptosporidiosis is mainly associated with C. parvum infections. The frequency of infections is varied and depends on the animals’ age and the place of breeding [24, 33, 34]. The youngest cattle up to the age of 8 weeks are the most susceptible to infection [4, 16, 35, 36]; therefore, the highest infection rate was observed in animals at that age [19]. The prevalence of C. parvum infections in the cattle population in Poland was estimated at 6.2%. As seen in our previous study the overall prevalence of C. parvum infections in cattle from Poland at the age of 1 day to 6 years was estimated at 5.1% but only animals below the age of 1 month were the major host for this parasite. In fact, C. bovis was the most frequently detected parasite species [15]. Furthermore, similar prevalence rate has been observed in cattle herds in north-eastern France (7.1%), Germany (9.2%) and Sweden (8.1–8.5%) [8, 18, 36, 37] and it was slightly higher than in Romania (5.4%) [38]. Higher prevalences of C. parvum in cattle have been reported outside Europe in Argentina (25.5%), Brazil (42.2%) and China (30–60%) [39,40,41].
It is believed that the animal production system (beef or dairy cattle) could have an impact on the frequency of infection [42, 43]. It is likely that different breeding practices related to animal production systems, for instance a continous calving season for milking cattle rather than periodic calving as in the case of meat breeds, could be considered factors associated with an increase in the number of infections [42]. In this study, C. parvum was more often detected in dairy Polish Black and White Holstein Friesian cattle than in meat or mixed dairy–meat animal breeds. This observation is consistent with previous findings that C. parvum infections occur more frequently in dairy cattle than in other breeds [42, 44]. Interestingly, it was also among dairy cattle where the most diverse population of C. parvum subtypes was found.
Based on the microsatellite sequence analysis of the GP60 gene of C. parvum strains isolated from Polish cattle, 11 subtypes representing the IIa and IId genetic families were detected. The six subtypes IIaA10G1R1, IIaA15G2R1, IIaA16G1R1b, IIaA17G1R1, IIaA17G2R1 and IIdA24G1c were previously identified in a bovine host in Poland [16]. Considering the analysed time-related changes on strain prevalence, infections caused by IIaA17G1R1 and IIaA17G2R1 dominate in cattle in Poland and these subtypes often jointly circulate in the same geographical area. In Europe, they have been detected in cattle with frequency varying from 0.4% of IIaA17G1R1 in Northern Ireland [35] to 14.5% in the case of IIaA17G2R1 in Italy [17]. However, they are not considered the dominating C. parvum subtypes in European cattle herds [33]. In contrast to the Polish situation, the most widespread subtype in livestock in Europe is IIaA15G2R1 [20, 23, 26, 43, 45]. It is worth noting that the IIaA15G2R1 strains together with IIaA16G1R1b constituted the second largest population of detected strains in cattle in Poland. Strains of the IIaA16G1R1b subtype of C. parvum as well as other closely related strains such as IIaA16G3R1 and IIaA16G2R1 have also been identified in livestock in Europe [33, 34]. In the group of IIaA16 variants in Poland, IIaA16G1R1b superabounded. Its presence has been revealed in cattle in Sweden [46] and in Serbia and Montenegro, where its occurrence was considerably heavier than that of other C. parvum strains [47]. In Europe, particularly in its south-eastern part, IIaA16G1R1 strains seem to be much more prevalent in cattle [3, 4, 48, 49].
In this study, five new C. parvum subtypes (A21R1, A18G3R1, A22G1R1, A21G1R1 and A14G2R1) from the IIa genetic family were detected. They had not been identified in calves in Poland prior to this. The strain IIaA18G3R1 has been previously found in livestock elsewhere in Europe [7, 35]. Its occasional presence has been reported in cattle in the Netherlands (0.8%), while a higher 5.7% prevalence rate has been observed in Spain [7, 22]. This strain was the most abundant in animals in Northern Ireland [35]. Interestingly, its common prevalence has been demonstrated in cattle and humans in Australia [50, 51]. In the case of C. parvum IIaA14G2R1, its prevalence in cattle in Europe has not exceeded 3.8% [7, 20, 23, 43]. In the group of newly detected strains in Poland is the one designated IIaA21R1. Surprisingly, elsewhere in Europe it has only been found in humans, these infections having been in Sweden and Norway [52]. The other two new subtypes IIaA22G1R1 and IIaA21G1R1 have been identified in cattle in Germany, Estonia, Sweden and Argentina [18, 20, 53, 54].
There was no relationship observed between infections caused by a specific parasite subtype and cattle age, and this observation is consistant with our previous finding [16]. However, a relationship between a subtype and its geographical prevalence in Poland was demonstrated. The IIaA17G1R1 strains were most frequently noted in cattle in southern Poland, while predominantly IIaA17G2R1 was found on farms located in southern, central and north-eastern Poland. Investigating epidemiology of Cryptosporidium infections in other species of farm ruminants in Poland, IIaA17G1R1 strain has also been occasionally detected in sheep [55]. Furthermore, the endemic presence of C. parvum IIaA16G1R1, IIaA19G1R1 and IIaA23G1R1 subtypes limited to one farm has been described by Del Coco et al. [53]. The authors did not indicate possible factors which could have contributed to this phenomenon. It is likely that other factors then age or animal breed could have an influence on diversity of detected C. parvum subtypes in cattle. Nevertheless, herd size, animal movement, farm management practices, or husbandry system could have an influence on regional subtype prevalence. However, based on the existing data, it would be difficult to determine their significance in the epidemiology of bovine cryptosporidiosis at subtype level.
Population dynamicity in C. parvum strains in cattle from Poland has been shown, as exemplified by the emergence of the IIlA19R3 subtype in the bovine host for the first time. In fact, the strains from the genetic family IIl rarely appear in Europe [4, 7, 47]. In the group of C. parvum strains which are disappearing from Poland are IIaA14G1R1, IIaA18G1R1c and IIaA19G1R1. Hitherto, the IIaA14G1R1 and IIaA19G1R1 subtypes have been identified in cattle in Estonia [54] and the Netherlands [7, 56] with frequencies of ≤ 7.9% (IIaA14G1R1) and ≤ 4.2% (IIaA19G1R1). In the case of IIaA19G1R1, it has usually been sporadically detected in cattle in Europe with infection prevalence not exeeding 3.9% [7, 23, 27, 54]. Elsewhere than Poland, IIaA18G1R1c strains were only found in diarrhoeic cattle in Sweden [46]. In Poland, C. parvum strains from the IId genetic family are the majority of those disappearing or found to occur endemically in particular geographical areas. Out of three IId subtypes circulating before 2014 in cattle, only one remained, IIdA24G1. It had not been identified in cattle in Europe, although it was present in humans [57,58,59]. However, a similar strain, IIdA24G1c, has been detected in Swedish cattle [46]. Nevertheless, the IIdA16G1b, IIdA17G1d, IIdA19G1, IIdA24G1c and IIdA26G1b bovine C. parvum subtypes from the IId genetic family were found in Europe with a frequency ranging from 1.1 to 15.3% [46].
As a final observation, the research findings of this study were limited by sample representativeness and lack of subtype identification for all detected C. parvum strains. Although the sampling scheme covered all Polish provinces, the number of sampled animals may not reflect their actual population size perfectly in each sampled region. Therefore, the findings for the variability of detected subtypes in a particular region may be biased and may not mirror the subtypes’ real transmission dynamics in cattle. A further shortcoming is that PCR amplification with GP60 primers was unsuccessful for 18 C. parvum strains, probably because of either the low quantity of Cryptosporidium DNA or low specificity of the primer’s sequences to the DNA sequence of the detected subtype. A greater number of identified strains could have had influence on the frequency of those strains’ detection and on the interpretation of subtype prevalence results. In particular this could be important for sporadically detected subtypes as it might have changed our perception of their prevalence.
Conclusions
The results of this study have enhanced our knowledge of the population genetics of C. parvum strains circulating over a period of 8 years in Polish cattle herds. Two strain subtypes predominated in cattle, i.e., IIaA17G1R1 and IIaA17G2R1, which over time were not displaced by other subtypes. Nevertheless, a certain strain population dynamicity was observed, which was associated with the disappearance of some strains (IIaA14G1R1, IIaA18G1R1c, IIaA19G1R1 and IIlA19R3) and emergence of new variants (IIaA18G3R1, IIaA14G2R1 and IIdA24G1) from the same genetic family in different geographical locations. The geographical change of strain prevalence from the western to the eastern part of Poland was evident for IIaA16G1R1b subtype. In the case of IIaA15G2R1 its prevalence range was geographically limited although it was circulating in livestock almost nationwide. Strains of the IId subtypes did not spread over time to other geographical locations suggesting their endemic prevalence. No age-dependent distribution of C. parvum subtypes was shown among the various cattle breeds being reared in Poland. Breed-dependent diversity of C. parvum subtypes was found, however, with the majority of subtypes being detected in HO cattle. The results of this study will also help in better understanding the subtype-related epidemiology of C. parvum infections in cattle and humans that could have originated from livestock, although they do not provide evidence for their possible transmission between hosts.
Materials and methods
Cattle faeces
In total, 1601 faecal samples were collected from healthy cattle at the age of 1 week to 4 months from 2014 to 2018 (Table 4). Farms (267) were randomly selected for visits in different locations across each of the 16 administrative provinces of Poland. The sampled animals were divided into three age groups: 1–4 weeks old (n = 527), > 4–8 weeks old (n = 464), and > 8–16 weeks old (n = 610) and were dairy varieties (Polish Black and White Holstein Friesian (HO), Jersey (JE), Polish Red and White Holstein Friesian (RW), Brown Swiss (BS), and Mixed exclusively dairy breed (MS)), meat varieties (Mixed exclusively meat breed (MM), Aberdeen Angus (AN), Charolaise (CH), Salers (SL), Limousie (LM), and Belgian Blue (BB)) and dairy–meat varieties (Mixed dairy–meat breed (MDM), Black and White lowland (NCB), Polish Red (RP), Montbeliarde (MO), Polish Black and White (ZB), and Simentaler (SM)). Freshly voided faecal samples of 10–15 g were placed individually into plastic containers, labelled and sent to the laboratory. For comparison studies analysing the time-related changes of geographical distribution of C. parvum subtypes in cattle herds in Poland, archive data on the occurrence in the Polish cattle population of specific subtypes were accessed. Those subtypes were A17G1R1, A17G2R1, A15G2R1, A16G1R1b, A10G1R1, A14G1R1, A16G3R1, A18G1R1c and A19G1R1 from IIa; A23G1, A24G1c and A22G1b from IId; and A19R3 from IIl genetic families. These strains were detected between 2010 and 2014 in different administrative provinces across Poland [16].
Detection of C. parvum in cattle and subtype identification
Samples were analysed using molecular methods according to previously described protocols [15]. Briefly, parasite genomic DNA was extracted from 0.1 g of faeces using a modified alkali wash and heat lysis method [15, 60]. Identification of C. parvum was performed using a PCR with primers targeting the 18 SSU rRNA locus [61] followed by a restriction fragment length polymorphism (RFLP) analysis of the obtained 849 bp amplicons [62, 63]. Extracts of DNA positive for C. parvum were subsequently used for subtyping of parasite strains by two nested GP60-PCR assays [64, 65] generating amplicons of approximately 800 bp or 400 bp respectively. Briefly, PCR reactions consisted of the following pre-mixed reagents: 300 µM of each of the four dNTPs, MgCl2 at either 1.5 or 3 mM, 1.25 U of KAPA Taq EXtra DNA polymerase in 1 × PCR buffer (Kapa Biosystems), 20 µg BSA (Thermo ScientificTM), and molecular grade water up to 50 µl reaction volume. Primers were used at the concentration of 0.1 µM (GP60 assays) or 0.5 µM each (18 SSU rRNA assay). Five or two µl of sample DNA or PCR products were the templates for primary and secondary amplifications respectively. Depending on the assay, the temperature profile was consisting of initial denaturation performed at 94 °C for 3–5 min, then 35 cycles of denaturation at 94 °C for 45 s., annealing at 55 °C (18SSU assay) or 50 °C /52 °C (GP60 assays) for 45 s., and extension at 72 °C for 1 min. A final extension step (72 °C for 7–10 min. depending on the assay) was included followed by a soak at 4 °C. All reactions were performed in a Biometra thermocycler (TProfessional BASIC). For each set of samples, the appropriate positive and negative controls were included during the nucleic acid extraction and PCR analyses. For restriction fragment analysis, 4 μl of nested-PCR product was digested in a total reaction volume of 10 µl consisting of 1 µl NdeI or MboII (Thermo ScientificTM) and the appropriate FastDigest reaction buffer at 37 °C for 1 h followed by 65 °C for 5 min. PCR products and their restriction fragments were analysed in 2.5% agarose gels stained with SimplySafe (Eurx).
The GP60 amplicons were excised from the agarose gel, purified and directly sequenced in both directions using the ABI Prism BigDye Terminator v3.1 Cycle sequencing kit on an ABI 3730XL DNA sequencer (Life Technologies, Carlsbad, CA, USA) at the Genomed S.A. sequencing service. Particular subtypes were identified based on trinucleotide repeats present in the analysed sequences. A single representative sequences for each subtype group sharing mutual 100% sequence similarity as well as all sequences that did not show 100% genetic resemblance within particular subtype group were deposited in the GenBank under the accession numbers OM836436—OM836449. The sequence similarity of detected strains was assessed by the maximum likelihood method with a Tamura-Nei parameter model using MEGA version 7.0.9 [66]. The reliability assessment of a topology (bootstrap) of the phylogenetic tree was performed at 1,000 replicates.
Statistical analyses
The significance of differences in the frequency of C. parvum infections between age groups of animals was determined by a chi-square (χ2) test. The frequency of parasite occurrence in cattle in different Polish regions (provinces) was analysed using a Kruskal–Wallis Chi-squared test. The relationship between the occurrence of C. parvum infection and breed expressed as true prevalence estimate (TPI) was evaluated based on Bayesian posterior probability (POPR) [67]. Estimates for each breed were shown for minimal and maximal POPR thresholds. A difference was regarded as statistically different when POPR was either smaller than 0.05 or grater than 0.95. Data on the sensitivity and specificity of the molecular method employed for parasite detection was included in the model to assess its influence on the estimates [68].
Concluding the statistical work, the χ2 test was also employed to show the relationships between C. parvum subtypes and their occurrence in calves of different breeds and ages by group and between subtypes and their geographical distribution in Poland. The frequency of occurrence of C. parvum subtypes in the studied cattle population was assessed by ANOVA. The calculations were performed with Statgraphics Centurion v. XVI (Statpoint Technologies, Warrenton, VA, USA) and R software v. 4.1.1 [69].
Availability of data and materials
The sequences of GP60 gene of C. parvum strains were deposited into NCBI GenBank under accesion numbers OM836436—OM836449.
Abbreviations
- C. parvum :
-
Cryptosporidium parvum
- PCR:
-
Polymerase chain reaction
- 18SSU rRNA:
-
Small subunit rRNA gene
- RFLP:
-
Restriction fragment length polymorphism
- GP60:
-
60-KDa glycoprotein gene
References
De Graaf DC, Vanopdenbosch E, Ortega-Mora LM, Abbassi H, Peeters JE. A review of the importance of cryptosporidiosis in farm animals. Int J Parasitol. 1999;29:1269–87.
Thomson S, Hamilton CA, Hope JC, Katzer F, Mabbott NA, Morrison LJ, et al. Bovine cryptosporidiosis: impact, host-parasite interaction and control strategies. Vet Res. 2017;48:42. https://doi.org/10.1186/s13567-017-0447-0.
Plutzer J, Karanis P. Genotype and subtype analyses of Cryptosporidium isolates from cattle in Hungary. Vet Parasitol. 2007;146:357–62.
Soba B, Logar J. Genetic classification of Cryptosporidium isolates from humans and calves in Slovenia. Parasitology. 2008;135:1263–70.
Cui Z, Wang R, Huang J, Wang H, Zhao J, Luo N, et al. Cryptosporidiosis caused by Cryptosporidium parvum subtype IIdA15G1 at a dairy farm in Northwestern China. Parasit Vectors. 2014;7:529.
Li N, Wang R, Cai MW, Feng Y, Xiao L. Outbreak of cryptosporidiosis due to Cryptosporidium parvum subtype IIdA19G1 in neonatal calves on a dairy farm in China. Int J Parasitol. 2019;49:569–77.
Wielinga PR, de Vries A, van der Goot TH, Mank T, Mars MH, Kortbeek LM, et al. Molecular epidemiology of Cryptosporidium in humans and cattle in The Netherlands. Int J Parasitol. 2008;38:809–17.
Björkman C, Lindström L, Oweson C, Ahola H, Troell K, Axén C. Cryptosporidium infections in suckler herd beef calves. Parasitology. 2015;142:1108–14.
Santín M, Trout JM, Xiao L, Zhou L, Greiner E, Fayer R. Prevalence and age-related variation of Cryptosporidium species and genotypes in dairy calves. Vet Parasitol. 2004;21(122):103–17. https://doi.org/10.1016/j.vetpar.2004.03.020.
Budu-Amoako E, Greenwood SJ, Dixon BR, Barkema HW, McClure JT. Occurrence of Cryptosporidium and Giardia on beef farms and water sources within the vicinity of the farms on Prince Edward Island, Canada. Vet Parasitol. 2012;184:1–9.
Amer S, Honma H, Ikarashi M, Tada C, Fukuda Y, Suyama Y, et al. Cryptosporidium genotypes and subtypes in dairy calves in Egypt. Vet Parasitol. 2010;11(169):382–6.
Samra AN, Jori F, Cacciò SM, Frean J, Poonsamy B, Thompson PN. Cryptosporidium genotypes in children and calves living at the wildlife or livestock interface of the Kruger National Park, South Africa. Onderstepoort J Vet Res. 2016;83:a1024. https://doi.org/10.4102/ojvr.v83i1.1024.
Uga S, Matsuo J, Kono E, Kimura K, Inoue M, Rai SK, et al. Prevalence of Cryptosporidium parvum infection and pattern of oocyst shedding in calves in Japan. Vet Parasitol. 2000;94:27–32.
Matsuura Y, Matsubayashi M, Nukata S, Shibahara T, Ayukawa O, Kondo Y, et al. Report of fatal mixed infection with Cryptosporidium parvum and Giardia intestinalis in neonatal calves. Acta Parasitol. 2017;62:214–20.
Rzeżutka A, Kaupke A. Occurrence and molecular identification of Cryptosporidium species isolated from cattle in Poland. Vet Parasitol. 2013;196:301–6.
Kaupke A, Rzeżutka A. Emergence of novel subtypes of Cryptosporidium parvum in calves in Poland. Parasitol Res. 2015;114:4709–16.
Duranti A, Cacciò SM, Pozio E, Di Egidio A, De Curtis M, Battisti A, et al. Risk factors associated with Cryptosporidium parvum infection in cattle. Zoonoses Public Health. 2009;56:176–82.
Silverlås C, Näslund K, Björkman C, Mattsson JG. Molecular characterisation of Cryptosporidium isolates from Swedish dairy cattle in relation to age, diarrhoea and region. Vet Parasitol. 2010;169:289–95.
Díaz P, Varcasia A, Pipia AP, Tamponi C, Sanna G, Prieto A, et al. Molecular characterisation and risk factor analysis of Cryptosporidium spp. in calves from Italy. Parasitol Res. 2018;117:3081–90.
Broglia A, Reckinger S, Cacció SM, Nöckler K. Distribution of Cryptosporidium parvum subtypes in calves in Germany. Vet Parasitol. 2008;154:8–13.
Vieira PM, Mederle N, Lobo ML, Imre K, Mederle O, Xiao L, et al. Molecular characterisation of Cryptosporidium (Apicomplexa) in children and cattle in Romania. Folia Parasitol. 2015;62:002. https://doi.org/10.14411/fp.2015.002.
Quílez J, Torres E, Chalmers RM, Robinson G, Del Cacho E, Sanchez-Acedo C. Cryptosporidium species and subtype analysis from dairy calves in Spain. Parasitology. 2008;135:1613–20.
Brook EJ, Anthony Hart C, French NP, Christley RM. Molecular epidemiology of Cryptosporidium subtypes in cattle in England. Vet J. 2009;179:378–82.
Kaupke A, Rzeżutka A. Epidemiologia inwazji Cryptosporidium parvum u zwierząt gospodarskich i wolno żyjących. Med Weter. 2017;73:387–94.
Holzhausen I, Lendner M, Göhring F, Steinhöfel I, Daugschies A. Distribution of Cryptosporidium parvum gp60 subtypes in calf herds of Saxony, Germany. Parasitol Res. 2019;118:1549–58.
Lichtmannsperger K, Hinney B, Joachim A, Wittek T. Molecular characterization of Giardia intestinalis and Cryptosporidium parvum from calves with diarrhoea in Austria and evaluation of point-of-care tests. Comp Immunol Microbiol Infect Dis. 2019;66:101333. https://doi.org/10.1016/j.cimid.2019.101333.
Mammeri M, Chevillot A, Chenafi I, Thomas M, Julien Ch, Vallée I. Polack B. Follet J. Adjou KT. Molecular characterization of Cryptosporidium isolates from diarrheal dairy calves in France. Vet Parasitol Reg Stud Reports 2019;18,100323. https://doi.org/10.1016/j.vprsr.2019.100323.
Cacciò SM, Chalmers RM. Human cryptosporidiosis in Europe. Clin Microbiol Infect. 2016;22:471–80.
Starkey SR, Johnson AL, Ziegler PE, Mohammed HO. An outbreak of cryptosporidiosis among alpaca crias and their human caregivers. J Am Vet Med Assoc. 2007;231:1562–7.
Mattsson JG, Insulander M, Lebbad M, Björkman C, Svenungsson B. Molecular typing of Cryptosporidium parvum associated with a diarrhoea outbreak identifies two sources of exposure. Epidemiol Infect. 2008;136:1147–52.
Lange H, Johansen OH, Vold L, Robertson LJ, Anthonisen IL, Nygard K. Second outbreak of infection with a rare Cryptosporidium parvum genotype in schoolchildren associated with contact with lambs/goat kids at a holiday farm in Norway. Epidemiol Infect. 2014;142:2105–13.
Galuppi R, Piva S, Castagnetti C, Sarli G, Iacono E, Fioravanti ML, et al. Cryptosporidium parvum: From foal to veterinary students. Vet Parasitol. 2016;219:53–6.
Imre K, Dărăbuș G. Distribution of Cryptosporidium species, genotypes and C. parvum subtypes in cattle in European countries. Sci Parasitol. 2011;12:1–9.
Avendaño VC, Amaya MA. Molecular characterization of Cryptosporidium parvum and Cryptosporidium hominis GP60 subtypes worldwide. Rev MVZ Córdoba. 2017;22:6339–49.
Thompson HP, Dooley JS, Kenny J, McCoy M, Lowery CJ, Moore JE, et al. Genotypes and subtypes of Cryptosporidium spp. in neonatal calves in Northern Ireland. Parasitol Res. 2007;100:619–24.
Díaz P, Navarro E, Remesar S, García-Dios D, Martínez-Calabuig N, Prieto A, et al. The age-related Cryptosporidium species distribution in asymptomatic cattle from North-Western Spain. Animals. 2021;11:256. https://doi.org/10.3390/ani11020256.
Heckler RP, Borges DG, Bacha FB, Onizuka MK, Le T, Neves JP, et al. First genetic identification of Cryptosporidium parvum subtype IIaA14G2R1in beef cattle in Brazil. Prev Vet Med. 2015;121:391–4.
Imre K, Lobo LM, Matos O, Popescu C, Genchi C, Dărăbuş G. Molecular characterisation of Cryptosporidium isolates from pre-weaned calves in Romania: is there an actual risk of zoonotic infections? Vet Parasitol. 2011;181:321–4.
Toledo RD, Martins FD, Ferreira FP, de Almeida JC, Ogawa L, Dos Santos HL, et al. Cryptosporidium spp. and Giardia spp. in feces and water and the associated exposure factors on dairy farms. PLoS One. 2017;12:e0175311. https://doi.org/10.1371/journal.pone.0175311.
Lombardelli JA, Tomazic ML, Schnittger L, Tiranti KI. Prevalence of Cryptosporidium parvum in dairy calves and GP60 subtyping of diarrheic calves in central Argentina. Parasitol Res. 2019;118:2079–86.
Zhang Z, Hu S, Zhao W, Guo Y, Li N, Zheng Z, et al. Population structure and geographical segregation of Cryptosporidium parvum IId subtypes in cattle in China. Parasit Vectors. 2020;13:425. https://doi.org/10.1186/s13071-020-04303-y.
Kváč M, Kouba M, Vítovec J. Age-related and housing-dependence of Cryptosporidium infection of calves from dairy and beef herds in South Bohemia, Czech Republic. Vet Parasitol. 2006;137:202–9.
Geurden T, Thomas P, Casaert S, Vercruysse J, Claerebout E. Prevalence and molecular characterisation of Cryptosporidium and Giardia in lambs and goat kids in Belgium. Vet Parasitol. 2008;155:142–5.
Dixon B, Parrington L, Cook A, Pintar K, Pollari F, Kelton D, et al. The potential for zoonotic transmission of Giardia duodenalis and Cryptosporidium spp. from beef and dairy cattle in Ontario, Canada. Vet Parasitol. 2011;10(175):20–6.
Rieux A, Paraud C, Pors I, Chartier C. Molecular characterization of Cryptosporidium isolates from pre-weaned calves in western France in relation to age. Vet Parasitol. 2013;197:7–12.
Silverlås C, Bosaeus-Reineck H, Näslund K, Björkman C. Is there a need for improved Cryptosporidium diagnostics in Swedish calves? Int J Parasitol. 2013;43:155–61.
Mišic Z, Abe N. Subtype analysis of Cryptosporidium parvum isolates from calves on farms around Belgrade, Serbia and Montenegro, using the 60 kDa glycoprotein gene sequences. Parasitology. 2007;134:351–8.
Imre K, Matos O, Dărăbus G, Mederle N, Oprescu I, Morariu S, et al. First genetic identification of Cryptosporidium spp. in cattle in Romania. Lucr Şt Med Vet Timişoara. 2009;52:26–30.
Imre K, Dărăbuş G, Mederle N, Oprescu I, Morariu S, Ilie M, et al. Intraspecific characterization of some Cryptosporidium parvum isolates from calves and lambs in Western Romania using molecular techniques. Sci Parasitol. 2010;11:47–50.
O'Brien E, McInnes L, Ryan U. Cryptosporidium GP60 genotypes from humans and domesticated animals in Australia, North America and Europe. Exp Parasitol. 2008;118:118–21.
Ng JS, Pingault N, Gibbs R, Koehler A, Ryan U. Molecular characterisation of Cryptosporidium outbreaks in Western and South Australia. Exp Parasitol. 2010;125:325–8.
Insulander M, Silverlås C, Lebbad M, Karlsson L, Mattsson J, Svenungsson B. Molecular epidemiology and clinical manifestations of human cryptosporidiosis in Sweden. Epidemiol Infect. 2013;141:1009–20.
Del Coco VF, Cordoba MA, Bilbao G, de Almeida Castro AP, Basualdo JA, Fayer R, et al. Cryptosporidium parvum GP60 subtypes in dairy cattle from Buenos Aires, Argentina. Res Vet Sci. 2014;96:311–4.
Santoro A, Dorbek-Kolin E, Jeremejeva J, Tummeleht L, Orro T, Jokelainen P, et al. Molecular epidemiology of Cryptosporidium spp. in calves in Estonia: high prevalence of Cryptosporidium parvum shedding and 10 subtypes identified. Parasitology. 2019;146:261–7.
Kaupke A, Michalski MM, Rzeżutka A. Diversity of Cryptosporidium species occurring in sheep and goat breeds reared in Poland. Parasitol Res. 2017;116:871–9.
Pinto P, Ribeiro CA, Hoque S, Hammouma O, Leruste H, Détriché S, et al. Cross-border investigations on the prevalence and transmission dynamics of Cryptosporidium species in dairy cattle farms in Western mainland Europe. Microorganisms. 2021;9:2394. https://doi.org/10.3390/microorganisms9112394.
Chalmers RM, Smith RP, Hadfield SJ, Elwin K, Giles M. Zoonotic linkage and variation in Cryptosporidium parvum from patients in the United Kingdom. Parasitol Res. 2011;108:1321–5.
Gherasim A, Lebbad M, Insulander M, Decraene V, Kling A, Hjertqvist M, et al. Two geographically separated food-borne outbreaks in Sweden linked by an unusual Cryptosporidium parvum subtype, October 2010. Euro Surveill. 2012;17:20318. https://doi.org/10.2807/ese.17.46.20318-en.
Hadfield SJ, Pachebat JA, Swain MT, Robinson G, Cameron SJ, Alexander J, et al. Generation of whole genome sequences of new Cryptosporidium hominis and Cryptosporidium parvum isolates directly from stool samples. BMC Genomics. 2015;16:650. https://doi.org/10.1186/s12864-015-1805-9.
Millar C, Moore J, Lowery C, McCorry K, Dooley J. Successful PCR amplification of genomic DNA from Cryptosporidium parvum oocysts extracted from a human faecal sample: a rapid and simple method suited for outbreak analysis. Int J Hyg Environ Health. 2001;204:191–4.
Xiao L, Escalante L, Yang C, Sulaiman I, Escalante AA, Montali RJ, et al. Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl Environ Microbiol. 1999;65:1578–83.
Feng Y, Ortega Y, He G, Das P, Xu M, Zhang X, et al. Wide geographic distribution of Cryptosporidium bovis and the deer-like genotype in bovines. Vet Parasitol. 2007;144:1–9.
Zintl A, Neville D, Maguire D, Fanning S, Mulcahy G, Smith HV, et al. Prevalence of Cryptosporidium species in intensively farmed pigs in Ireland. Parasitology. 2007;134:1575–82.
Glaberman S, Moore JE, Lowery CJ, Chalmers RM, Sulaiman I, Elwin K, et al. Three drinking-water-associated cryptosporidiosis outbreaks, Northern Ireland. Emerg Infect Dis. 2002;8:631–3.
Sulaiman IM, Lal AA, Xiao L. Molecular phylogeny and evolutionary relationships of Cryptosporidium parasites at the actin locus. J Parasitol. 2002;88:388–94.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. https://doi.org/10.1093/molbev/msw054.
Mweu MM, Toft N, Katholm J, Nielsen SS. Evaluation of two herd-level diagnostic tests for Streptococcus agalactiae using a latent class approach. Vet Microbiol. 2012;159:181–6.
Mirhashemi EM, Zintl A, Grant T, Lucy FE, Mulcahy G, De Waal T. Comparison of diagnostic techniques for the detection of Cryptosporidium oocysts in animal samples. Exp Parasitol. 2015;151-152:14–20.
R Core Team R. A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2021. https://www.R-project.org
Acknowledgements
The authors are thankful to all the veterinary surgeons who helped with sample collection, and are grateful to Zbigniew Osiński for statistical analysis and interpretation of some results and Dorota Fijoł for technical assistance.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceptualization: AK, AR; Methodology: AK; Investigation, results analysis: AK, AR; Assisted in drafting of the manuscript: AK; Writing—original draft preparation, manuscript review and editing, supervision: AR. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study did not require approval of Ethics Committee. However, freshly voided faeces was collected during routine veterinary practice in adherence to international guidelines of animal care. No clinical interventions were performed. The owners of the cattle included in this study provided informed consent to participate.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kaupke, A., Rzeżutka, A. Population genetics of Cryptosporidium parvum subtypes in cattle in Poland: the geographical change of strain prevalence and circulation over time. BMC Vet Res 18, 263 (2022). https://doi.org/10.1186/s12917-022-03328-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-022-03328-y