
Abstract
Background
Porcine reproductive and respiratory syndrome (PRRS) is one of the most expensive diseases of modern swine production & results in annual economic losses and cost the industry over 600 million USD in U.S. alone and billions of dollars worldwide. Two atypical PRRS cases were observed in 2013 and 2014 characterized by late-term abortion, fever and sudden increase in sow mortality which persisted for a prolonged period of time.
Methods
Lungs, lymph nodes and other samples were collected for disease investigation. Sequencing of the viral envelope glycoprotein (ORF5) and nucleocapsid protein (ORF7) of PRRSV was done using the BigDye Terminator v3.1 cycle sequencing kit chemistry. The phylogenetic tree was constructed by using the Maximum Likelihood method, generated by Mega 6.06®.
Results
Analysis of the ORF5 and ORF7 showed high degree of sequence homology to PRRSV parent vaccine strain VR-2332, RespPRRSV and other mutant/chimeric virus strains.
Conclusions
Our study suggests that recombination events between vaccine strains and field isolates may contribute to PRRSV virulence in the field.
Similar content being viewed by others

Background
Porcine reproductive and respiratory Syndrome (PRRS) is an economically important viral disease that is easily transmitted through direct contact to susceptible pigs and vertically to foetuses. The disease is also known as Mystery Swine Disease, Blue Ear Disease, Porcine Endemic Abortion & Respiratory Syndrome (PEARS) and Swine Infertility Respiratory Syndrome (SIRS) [1, 2]. It is known as one of the most expensive disease of modern swine production. A porcine reproductive and respiratory syndrome virus (PRRSV) outbreak can devastate a herd and determining the origin of the virus can be impossible. PRRS is characterized by an acute viral infection of the porcine macrophage that leads to an immunologically altered state. In extreme cases, respiratory distress, metabolic dysregulation and neuronal involvement result in significant mortality within days to weeks of experimental inoculation with highly pathogenic isolates [3–5]. The virus can also reappear in farms that have taken great lengths to eliminate the virus. Endemic disease from emerging and re-emerging PRRSV results in estimated annual economic losses and the virus is estimated to cost the industry over 600 million USD, or 1.5 million USD per day in the U.S. economy alone [6].
Since its emergence, much has been studied and learned about the virus. It was first detected in North America and reported in 1987 [1]. The virus was then subsequently isolated in Europe in 1990 [7]. Since then it has spread rapidly to Asia and throughout the world. The porcine reproductive & respiratory syndrome virus (PRRSV), the causative agent for the syndrome, is a positive-sense single stranded RNA virus, belonging to the family Arteriviridae of the order Nidovirales, and genus Arterivirus [8, 9].
The PRRSV genome organization is similar to other arterivirus and is approximately 15 kilobases in length. There are 10 open reading frames (ORFs), ORF1a and ORF1b encoding polyproteins that are processed into 14 non-structural proteins (nsp) by viral proteases within the virus genome [10]. The glycosylated membrane associated minor structural proteins GP (2a), GP3 and GP4, respectively are encoded by ORF2a, ORF3 and ORF4 [11]. ORF2b encodes 2b protein, a non-glycosylated structural protein which is virion associated and the principal product of ORF2 [12]. Three major structural proteins, GP5, M and N protein within the virus genome are encoded by ORF5, ORF6 and ORF7, respectively. GP5a, which is referred to as ORF5a protein, is a novel structural protein encoded by an alternative ORF of the subgenomic mRNA encoding GP5 and is incorporated into the virion [13, 14].
Based on genetic characterization, there exist two related but antigenically and genetically distinguishable major genotypes with over 50% RNA sequence variation; the European strain (EU genotype, Type 1, with Lelystad virus as the prototype) representing the viruses predominating in Europe and the North American strain (NA genotype, Type 2, with VR-2332 as the prototype) originally and mostly found in North America [15]. Both genotypes have been described to be evolving independently in Europe and North America and the co-existence of both genotypes has been increasingly evident in several countries, including Malaysia, Thailand, Korea and China [16–20]. Most recently, a variant of genotype 2 also known as highly pathogenic strain of PRRSV, genetically characterized by a unique discontinuous deletion of 30 amino acids (aa) in the non structural protein (Nsp2) of the North American strains was confirmed by the Office International Des Epizooties (OIE) and the Food and Agricultural Organization (FAO) as the causative agent for the severe “high fever” disease designated as the highly pathogenic strain of PRRSV in Asia. Because of its economic significance, a great deal of resource has been invested to research the virus and in developing effective prevention and control strategies. But protocols providing consistent success have been elusive due to the high rate of genetic change and antigenic variability [2, 21–24].
Situation in South East Asia
Throughout Asia, PRRS outbreaks were reported in many countries between the late 1980s and early 1990s [9]. The highly pathogenic PRRS (HP-PRRS) which emerged in China in 2006 has spread to South East Asian countries since 2007 [25]. The highly pathogenic PRRS was reported in Vietnam in March 2007 [26], Laos in June 2010 [27], Thailand in 2010 [28], Myanmar in February 2011 [29], Cambodia in August 2010 [30], Philippines in August 2010 [30]. Transboundary spread of HP-PRRSV from southern China to South East Asia suggests that biosecurity failures have occurred, including failure to control animal movements and trading among neighboring countries at borders [31, 32] (Fig. 1).

Is Malaysia at risk of HP-PRRSV? Since the disease started in China, it has quickly spread to the surrounding countries in South East Asia. Malaysia is constantly at risk and threat after Thailand reported its first HP-PRRS in 2010
Situation in Malaysia
In Malaysia, a syndrome very similar to PRRSV has been recorded in various pig farms as early as 1995 [33]. A serological survey done in about 100 farms in major pig rearing states in the country showed that the pigs found in 93 out of the 100 farms had serological evidence of infection. Subsequently a study done in 2008 showed that 94% of the farms and 83.4% of the pigs were tested positive for PRRSV antibodies [34]. In 2012, another local study documented 89.2% sero-prevalence out of 120 sera collected from 12 non-PRRS vaccinated farms in 6 states. In the same study, 27 tissue samples were collected from 11 farms [35]. Twelve of the tissue samples were positive for PRRSV with all positive for US strains in the selected pig farms. The study concluded that there are more US strains in the selected pig farms, however, it should be noted that there were limitations as the number of samples studied were too small. In 2013, another study conducted in Malaysia concluded that both EU and NA strains are present in Malaysian [36].
Farming situation in Malaysia
There is approximately 772 farms in Malaysia with 565 farms in West Malaysia with an approximate total no of sows of 0.17 million of which 0.14 million is located in West Malaysia. The estimated total ex farm value of the swine industry in Malaysia is around 2.2 billion ringgit. Similar to the farming situation in other countries, the farming industry in Malaysia has moved on from backyard to industrialized farming systems, with many small farms that has shut down; merged or bought over by bigger players in the industry which explains the reason for the number of farms that have reduced but the farm sizes that are increasing. The farms are mostly open house, farrow to finish and single site, about 10% of the farms use closed house system. The swine respiratory health status varies from state to state and from farm to farm as well. Selangor (located in Central Malaysia) and Penang (located in North Malaysia) are highly urbanized states with limited land space; therefore the farms are close to each other. Johor (located in South Malaysia and borders with Singapore)—these farms used to export to Singapore and are more established. Perak is located between central and north Malaysia which used to be a mining area; the pig farms are located next to lakes and ponds. Sabah and Sarawak which is located in East Malaysia have more land and space, thus, in theory there are less issues.
Current study
Between 2013–2014, 22 diagnostic cases were received for diagnostic investigation. Seven out of 22 cases received were positive. Six out of the seven cases were positive for NA strain while one case was positive for both EU and NA strain. Among all those cases that were received, there were two atypical cases observed in 2013 and subsequently in 2014 in which high mortality rates were observed in the farm.
Case 1: Location: East Malaysia; Year: 2013
History & clinical signs
From November through December of 2012, high mortality was observed in a pig farm in East Malaysia with its return/repeat service increasing by 30%. The piglets were weak and runt. It was also evident that the disease was spreading along to pig farms located in close proximity. About 2–3 months later, there were occurrence of problems in weaners showing signs of Edema and Classical Swine Fever virus and other bacterial infections. At that point, veterinarians were called to assist to collect samples and investigate the disease. Upon tracing back the case history and through differential diagnosis, there were adequate reasons to suspect the case as a potential high fever PRRSV (Fig. 2).

Case 1 (Sar01/2013) East Malaysia. a, b, c, d: At the time of sample collection, the case looked like Edema and the animals had secondary bacterial infections. e, f: The virus had spread to the neighbouring farms and showed similar clinical signs. 3 days old piglets looked weak, runt and emaciated
Case 2: Location: Central Malaysia, Year: 2014
History & Clinical Signs
In February 2014, a farrow-finish 300 sow herd reported an outbreak of late-term abortion, 30% of repeat to estrus, and more than 50% of pre-weaning mortality. The sow herd had been regularly vaccinated with Aujeszky’s vaccine, swine fever and PRRS MLV. Mortality of grower & finisher were increased with porcine respiratory disease complex. One week after a schedule blanket vaccination in sow herd with PRRS MLV, there were more than 50% of gilt and first parity sow where sudden death near to term with hyperemia and pyrexia, and 20% of late-term abortion. Umbilical hemorrhage was observed in some cases. Piglet showed ill thrift with periocular oedema and conjunctivitis. Weaners exhibited dyspnea and lethargy and mortality rose to more than 80% (Fig. 3).

Case 2 (Sel01/2014) Central Malaysia. a and b: The piglets are weak, runt and chilled. They are clustering together to stay warm. c: Sudden death detected in sow with hyperemia and pyrexic

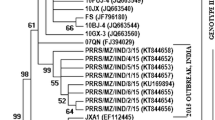
Molecular Phylogenetic analysis of the ORF5 gene of Case 1 (Sar01/2013) & Case 2 (Sel01/2014) from East Malaysia. (Source of Map: http://aseanup.com/free-maps-asean-southeast-asia/)

Molecular Phylogenetic analysis of the ORF7 gene of Case 1 (Sar01/2013) & Case 2 (Sel01/2014) from East Malaysia
In this current study, we compare the ORF5 and ORF7 gene sequence from Malaysia with the ORF5 and ORF7 gene isolates from other Asian countries to study the diversity of PRRSV in Malaysia which may help shed some light on the potential origin of PRRSVs in Malaysia. ORF5 encodes the major viral envelope glycoprotein (GP5), which is located on the surface of the virion. GP5 plays an important role in viral infectivity and contains important immunological domains associated with viral neutralization [13, 14]. Several peptide/protein motifs, such as signal peptides, trans-membrane regions, antigenic determinants and glycosylation sites have been widely used for analyzing genetic variation and the molecular epidemiology of PRRSV. The ORF7 encodes the nucleocapsid protein, the most abundant viral protein in virus-infected cells and the most immunodominant antigen in the pig immune response to PRRSV. ORF7 is, therefore a promising candidate for detection and diagnosis [10, 11].
Results and Discussion
Analysis of the nsp2 gene of Case 1 and Case 2
After various attempts and optimization strategies, the team did not manage to obtain any positive bands from Case 1 and Case 2 for the nsp2 gene despite getting a positive band from positive controls. It is highly suspected that the nsp2 regions of both cases are highly mutated and not amplifiable with known published primers that are readily available. The nsp2 of PRRSV is a multi-domain protein that has been shown to undergo remarkable genetic variation. From its three major domains [37–39], nsp2 is the most divergent protein between PRRSV types 1 and 2 [40, 41] and also between the pathogenic PRRSV 16244B and an attenuated vaccine and its parental strain VR2332 [40–42]. A large amount of data supports the theory that the middle section of the nsp2 protein is highly susceptible to mutation and tolerant to insertions and deletions, regardless of the pathogenic phenotype of the isolates [39–50]. In fact, reverse genetics experiments have mapped several non-essential viral replication regions in nsp2, including a large deletion of 402 nucleotides in the middle region of the gene [39, 51].
Analysis of the ORF5 gene (KU512850) of Case 1 (Sar01/2013) from East Malaysia
BLAST analysis of the ORF5 sequence derived from Case 1 (Sar01/2013) from East Malaysia (KU512850) on Genbank showed that the sequence had high similarities to PRRSV sequences from China (KR612142; KR018787), South Korea (KP317086) and USA (KT905092; KT904941). Based on the phylogenetic study, the ORF5 gene sequence derived from Case 1 (Sar01/2013) from East Malaysia (KU512850) clustered together with the ORF5 gene sequences PL97-1 from South Korea (AY585241), 4034-2-v-2008 from South Korea (FJ972733), MD-001 from Taiwan (AF121131) and RespPRRS MLV (AF066183), MLV RespPRRS AF159149 and ATCC VR-2332 U87392.3, the parent strain of the vaccine Ingelvac PRRS MLV. The sequence comparison showed that the nucleotide sequence of the ORF5 gene derived from Case 1 (Sar01/2013) from East Malaysia (KU512850) had 99.3% nucleotide sequence homology with RespPRRS MLV (AF066183) and RespPRRS MLV (AF159149), 99% nucleotide sequence homology with ATCC VR-2332 (U87392.3) the parent strain of the vaccine Ingelvac PRRS MLV and 98% nucleotide sequence homology with 4034-2-V-2008 (FJ972733) from South Korea.
Three mismatched amino acid changes were observed in position 3, 4 and 13 throughout the ORF5 gene in comparison to ATCC VR-2332 (U87392.3). Amino acid substitutions at positions 3 (Glutamate, E) was substituted with (Glycine, G); amino acid substitution at position 4 (Lysine, K) was substituted with (Glutamate, E) and amino acid substitution at position 13 (Arginine, R) was substituted with (Glutamine, Q) was observed in Case 1 from East Malaysia (KU512850) (Fig. 6).

Amino acid sequence alignment of the ORF5 gene of Case 1 (Sar01/2013) and Case 2 (Sel01/2014) from East and Central Malaysia. Identical sequences are doted. Boxed portions denote mutations/changes in the amino acid
Analysis of the ORF7 gene (KU512849) of Case 1 (Sar01/2013) from East Malaysia
BLAST analysis on Genbank shows that the ORF7 gene sequence (KU512849) of Case 1 from East Malaysia derived in this study is highly similar to PRRSV strain JN-HS from Shandong, China, 2008 (Accession No: HM016158). Further analysis by phylogenetic tree studying only selected sequences of the ORF7 gene (KU512849) of Case 1 (Sar01/2013) from East Malaysia with other highly similar sequences from Genbank showed that it clustered with sequences from China (HM016158, KM453699, KM453698), Vietnam (JQ860406, JQ860392, JQ860419, JQ860410) and Laos (JN626287). The phylogenetic tree also suggest that the ORF7 gene (KU512849) of Case 1 (Sar01/2013) from East Malaysia is a derivative of JX-AI from Jiangxi, China (EF112445) which is a representative strain of highly pathogenic PRRSV in China since 2006. The sequence comparison of the nucleotide sequence of ORF7 gene (KU512849) of Case 1 (Sar01/2013) from East Malaysia showed 97.8% sequence homology to to JN-HS from China, 96.7% sequence homology to JX-A1 from Jiangxi, China and 97.5% sequence homology to these sequences from China (HM016158, KM453699, KM453698), Vietnam (JQ860406, JQ860392, JQ860419, JQ860410) and Laos (JN626287). The high genetic similarity between Case 1 (Sar01/2013) and JX-A1 (EF112445) suggest that Case 1 (Sar01/2013) from East Malaysia may be potentially a first HP-PRRSV found in Malaysia. Multiple mismatched amino acid changes were observed in position 15, 46, 91, 109, 117 and 122 throughout the ORF7 gene of Case 1 (Sar01/2013) from East Malaysia in (KU512849) comparison to ATCC VR-2332 (U87392.3).
Analysis of the ORF5 gene (KU512851) of Case 2 (Sel01/2014) from Central Malaysia
BLAST analysis on Genbank showed that the ORF5 gene sequence derived in this study was highly similar to PRRSV isolate Shizuoka from Japan (Accession No: AB175704.1) and other isolates from America (KT894735.1, U34298.1, DQ477864.1, DQ477718.1). One of the sequence that is highly similar to the ORF5 gene of Case 2 (Sel01/2014) from Central Malaysia is a synthetic contruct clone of PRRSV. Based on the phylogenetic study, the ORF5 gene sequence derived from this study clustered together with the ORF5 gene sequences from Taiwan (AF121131), South Korea (FJ972733; AY585241); RespPRRS MLV (AF066183), MLV RespPRRS AF159149 and ATCC VR-2332 U87392.3, the parent strain of the vaccine Ingelvac PRRS MLV. Sequences for constructing the phylogenetic tree were selected based on genetic relatedness and completeness of sequences that were available on Genbank. The sequence comparison demonstrated that the nucleotide sequence of ORF5 gene derived from Case 2 (Sel01/2014), Central Malaysia (KU512851) has 89.5% sequence homology with sequence MD-001 (AF121131) from Taiwan. The sequence homology of the ORF5 gene from Case 2 (Sel01/2014), Central Malaysia (KU512851) with the vaccine strains were relatively lower than expected with 85.5% with RespPRRS MLV (AF066183), 85.5% with MLV RespPRRS AF159149 and 86% with ATCC VR-2332 U87392.3, the parent strain of the vaccine Ingelvac PRRS MLV. This is not surprising as it has been documented that PRRSV strains differ in virulence [22, 52–55] and vary genetically [56–59] suggesting that the ORF5 gene derived from Case 2 (Sel01/2014), Central Malaysia (KU512851) may be a derivative of the Ingelvac PRRS MLV vaccine, a possible recombinant of the vaccine virus and a wild-type virus, or a truly wild-type virus that is partially homologous to the original parent vaccine strain, VR2332, which may be still circulating in the field [55].
As expected that the ORF5 that encodes for the major envelope glycoprotein would have a high degree of mutation as it is known to be the most variable region [55], multiple mismatched amino acids were observed throughout the ORF5 gene of Case 2 (Sel01/2014), Central Malaysia (KU512851) in comparison to ATCC VR-2332 (U87392.3) (Fig. 4).
Analysis of the ORF7 gene (KU512848) of Case 2 (Sel01/2014) from Central Malaysia
BLAST analysis on Genbank of the ORF7 gene sequence from Case 2 (Sel01/2014), Central Malaysia (KU512848) derived in this study demonstrates high similarity to PRRSV strains from South Korea, China, Denmark and USA. Further analysis by phylogenetic tree studying only selected sequences of the ORF7 gene with other highly similar sequences from Genbank suggest that the ancestor for the sequence from this study is ATCCVR2332 which is the parent strain to Ingelvac PRRS MLV vaccine (Fig. 5). The phylogenetic tree also suggest that the ORF7 gene sequence derived from this study groups closely with other sequences from Genbank such as V7-HA-myc (FJ524376), RespPRRS MLV (AF066183), RVRp22 (KM386622), FJSD (KP998474), DK-2004-1-7-PI (KC862578), DK-1997-19407B (KC862576), YN-2011 (JX857698), pMLV/MN184-3UTR (FJ629371) and pMLV/MN184ORF5-6 (FJ629369); two of which are chimeric infectious Porcine reproductive and respiratory syndrome virus type 2 clone representing a background of strain Ingelvac PRRS MLV and 3′UTR of MN184. The sequence comparison confirmed that the nucleotide sequence of ORF7 gene derived from this study is 98.3% similar to ATCC VR-2332 which is the parent strain to Ingelvac PRRS MLV vaccine and has 100% similarity to other sequences as listed in Table 1; two of which are chimeric infectious PRRSV pMLV/MN184-3′UTR FJ629371 & pMLV/MN184-3′UTR FJ629371. Three mismatched amino acid changes were observed in position 49, 54 and 56 throughout the ORF7 gene of Case 2 (Sel01/2014), Central Malaysia (KU512848) in comparison to ATCC VR-2332 (U87392.3) (Fig. 7). At first glance, the four sequences derived from these two cases display a vast diversity in terms of geographical origin of the PRRS virus found in Malaysia. However, when looked closely, all four sequences seem to have one thing in common, they display high sequence homology to the modified live vaccine virus strain regardless of which geographical location the sequences are highly similar to.

Amino acid sequence alignment of the nucleocapsid protein (ORF7) gene of Case 1 (Sar01/2013) and Case 2 (Sel01/2014) from East and Central Malaysia. Identical sequences are doted. Boxed portions denote mutations/changes in the amino acid
Both cases displayed a high degree of mutation in both genes, ORF5 and ORF7. There are several possibilities that the ORF5 and ORF7 genes from Case 1 (Sar01/2013) and Case 2 (Sel01/2014) are derivatives of the Ingelvac PRRS MLV vaccine, a possible recombinant of the vaccine virus and a wild-type virus, or a truly wild-type virus that is highly homologous to the original parent vaccine strain, VR 2332. This theory was discussed by Opriessnig in 2002 [55].
Further to that, it has been reported that the use of synthetic porcine reproductive and respiratory syndrome virus strain confers unprecedented levels of heterologous protection because current vaccines do not provide sufficient levels of protection against divergent PRRSV strains circulating in the field, mainly due to the substantial variation of the viral genome [60]. However, based on the phylogenetic study and sequence comparison, our study suggest that these synthetic chimeric infectious PRRSV constructs are highly similar to the PRRSV that are causing high mortality rate in the farm in Case 2 (Sel01/2014) in Central Malaysia. Therefore, it is apparent that the rapid rate of PRRSV gene mutation remains a huge challenge for the practicality of such synthetic clone vaccines.
Conclusion
Over the years, there have been biased and mixed reports of the presence of PRRSV in Malaysia. Based on a collection of studies, it can be confirmed that both European and North American strains are present in Malaysia. However, over the years there have been more North American strains reported compared to European strains. Despite global and intensive use of MLV vaccine, repeated PRRS outbreaks continue due to constant genetic changes in field isolates. Rapid evolution due to high mutation rate has led to new generations of genetically and antigenically variable virus strains in the field. Vaccine strains or derivatives of vaccine strains may also induce disease in the field and persist in vaccinated pigs and spread to non-vaccinated pigs contributing to PRRSV virulence in the field and the inability for effective vaccination. As reported by many other researchers in other countries [55], the results from this study suggest that the virus that persisted in both farms is a product of a recombination event between vaccine strains and field isolates. For future studies, genetic and evolutionary analyses of full length genomes are important to delineate the degree of homology among PRRSVs and for effective vaccine design. We are unable to confirm the presence of highly pathogenic PRRSV in Malaysia. The high degree of nucleotide homology between the local Malaysian isolates with representative strains of highly pathogenic PRRSV strains from China suggest that a derivative of the highly pathogenic virus strain may be present in Malaysia.
Methods
Sampling
Two sets of samples from two atypical PRRS cases were sent for disease investigation. One of the sample was from Central Malaysia, Selangor (lungs and lymp nodes) and another from East Malaysia, Sarawak (brains and lungs). The organ samples were pooled for diagnostic testing.
Animals were humanely slaughtered for disease investigation purpose by professionally trained veterinarians. There was no experimental research done on the animals.
Nucleic acid extraction
Nucleic acid extraction was carried out on the pooled organ samples by using TRIsure® (Bioline®). 100–200 mg of tissue was collected and homogenized using a mortar and pestle and 1 mL of Phosphate Buffered Saline (PBS). 750 uL of cold TRIsure® was added to 250 uL of the homogenized samples in a microcentrifuge tube. The samples were then vortexed vigorously and incubated at room temperature for 5 min. 200 uL of cold chloroform was then added into the same microcentrifuge tube, vortexed vigorously and incubated at room temperature for 5 min. The microcentrifuge tube is then centrifuged at 12, 000 rpm for 15 min at 4 °C. The upper aqueous solution was then transferred to a new and clean microcentrifuge tube. 5 uL polyacryl carrier (Molecular Research Centre Inc) and 500 uL of cold isopropanol are then added and the microcentrifuges containing these reagents were incubated at room temperature for 10 min. After 10 min, centrifuge at 12, 000 rpm for 10 min at 4 °C. Decant the supernatant and wash pellet with 1 mL of 75% cold ethanol. Centrifuge at 12, 000 rpm for 5 min at 4 °C. The pellet was resuspended in 50 uL of TE buffer.
PCR amplification of PRRSV—ORF5 and ORF7 gene
The presence of PRRSV in the samples were assessed using a previously described reverse transcriptase nested PCR assay that amplifies a 241 bp nucleotide (European strain) and 337 bp nucleotide (North American strains) respectively [61, 62]. Three sets of primers were used. PLS: 5′-ATG GCC AGC CAG TCA ATC-3′; PLR: 5-TCG CCC TAA TTG AAT AGG TG-3′ [62–64] to reverse transcribes and amplifies a common site in the ORF 7 region of both strains. The nested primer sets for the North American and European Strains were P-US-s: 5′-AGT CCA GAG GCA AGG GAC CG-3′; P-US-as:5′-TCA ATC AGT GCC ATT CAC CAC-3′ and P-EU-s:5′-ATG ATA AAG TCC CAG CGC CAG CGC CAG-3′; P-EU-as:5′-CTG TAT GAG CAA CCG GCA GCA T-3′.
PCR amplification with ORF5 gene
The primer pairs used were ORF5-F: 5′-ATGTTGGGGAAGTGCTTGACC-3′ and ORF5-R: 5′-CTAGAGACGACCCCATTGTTCCGC-3′ [65].
PCR amplification with nsp2 gene
The primer pairs used were 2492-F: 5′-GRACTTCCTCARCTTCTTGC-3′ and 3160-R: 5′-TCGACGAGCTTAAAGACCAGA-3′ [51].
Sequence alignment & phylogenetic analysis of the ORF5 and ORF7 region
Virus sequences were derived from clinical samples sent to the laboratory for diagnostic investigation, with the permission of local veterinary authorities. Virus RNA was extracted using TRIzol reagent according to the manufacturer’s instructions. RNA was quantified by using spectrophotometer (SpectraMax® Plus 384, Molecular Devices). A 603 bp (ORF5) and 337 bp (ORF7) region of the was amplified by reverse transcriptase PCR. The PCR products were purified using the PCR clean-up gel extraction kit according to the manufacturer’s protocol (Macherey-Nagel, Germany). Sequencing of the complete genome of PCV2 was done in a commercial sequencing facility using the BigDye Terminator v3.1 cycle sequencing kit. After sequencing, a Basic Local Alignment Search Tool (BLAST) was performed as a preliminary measure to confirm that all samples were true PRRSV when compared with other sequences deposited in Genbank. The sequence editing and assembly were done by using CLC Workbench. At least one forward and one reverse primer were used to generate a consensus sequence for each gene, which were visually checked for errors prior to alignment using CLC Workbench. Multiple sequence alignments were done by using ClustalW. The phylogenetic tree was constructed by using the Maximum Likelihood method based on the Tamura-Nei model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree (s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algoritms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branches lengths measures in the number of substitutions per site. Positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in Mega 6 (Biodesign Institute, Tempe, Arizona). The sequence identity matrix data was generated with BioEdit Sequence Alignment Editor version 7.0.5.2 (Tom Hall, US). Sequences used for constructing the phylogenetic tree are listed in Table 1.
Nucleotide sequence accession numbers
The complete genomic sequences of the ORF5 and ORF7 gene reported in this paper were deposited into the GenBank database under accession numbers KU512848, KU512849, KU512850 and KU512851.
Abbreviations
- ORF:
-
Open reading frame
- PCR:
-
Polymerase chain reaction
- PRRSV:
-
Porcine reproductive and respiratory syndrome virus
References
Keffaber KK. Reproductive failure of unknown etiology. Am Assoc Swine Vet. 1989;1(2):1–9.
Xiao XL, Wu H, Yu YG, Cheng BZ, Yang XQ, Chen G, Liu DM, Li XF. Rapid detection of a highly virulent Chinese-type isolate of Porcine Reproductive and Respiratory Syndrome Virus by real-time reverse transcriptase PCR. J Virol Methods. 2008;149(1):49–55.
Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, Wang X, Yang X, Kang L, Sun M, Jin P, Wang S, Kitamura Y, Yan J, Gao GF. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One. 2007;2:e526.
Guo B, Lager KM, Henningson JN, Miller LC, Schlink SN, Kappes MA, Kehrli Jr ME, Brockmeier SL, Nicholson TL, Yang HC, Faaberg KS. Experimental infection of United States swine with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. Virology. 2013;435:372–84.
Kappes MA, Miller CL, Faaberg KS. Highly Divergent Strains of Porcine Reproductive and Respiratory Syndrome Virus Incorporate Multiple Isoforms of Nonstructural Protein 2 into Virions. J Virol. 2013;87(24):13456–65.
Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmerman JJ, Rotto HF, Yoder TK, Wang C, Yeske PE, Mowrer CL, Haley CA. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod. 2013;21(2):72–84.
Wensvoort G, Terpstra C, Pol JM, ter Laak EA, Bloemraad M, de Kluyver EP, Kragten C, van Buiten L, den Besten A, Wagenaar F, Broekhuijsen JM, Moonen PLJM, Zetstra T, de Boer EA, Tibben HJ, de Jong MF, van Veld P, Greenland GJR, Van’t Gennep JA, Voets M, Verheijden JHM, Braamskamp J. Mystery swine disease in the Netherlands: the isolation of Lelystad virus. Vet Q. 1991;13:121–30.
Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629–33.
Zimmerman JJ, Yoon KJ, Pirtle EC, Wills RW, Sanderson TJ, McGinley MJ. Studies of porcine reproductive and respiratory syndrome (PRRS) virus infection in avian species. Vet Microbiol. 1997;55:329–36.
Fang Y, Snijder EJ. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 2010;154:61–76.
Music N, Gagnon CA. The role of porcine reproductive and respiratory syndrome (PRRS) virus structural and non-structural proteins in virus pathogenesis Anim. Health Res Rev. 2010;11:135–63.
Wu WH, Fang Y, Farwell R, Steffen-Bien M, Rowland RR, Christopher-Hennings J, Nelson EA. A 10-kDa structural protein of porcine reproductive and respiratory syndrome virus encoded by ORF2b. Virology. 2001;287:183–91.
Firth AE, Zevenhoven-Dobbe JC, Wills NM, Go YY, Balasuriya UB, Atkins JF, Snijder EJ, Posthuma CC. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J Gen Virol. 2011;92:1097–106.
Johnson CR, Griggs TF, Gnanandarajah J, Murtaugh MP. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J Gen Virol. 2011;92:1107–16.
Allende R, Lewis TL, Lu Z, Rock DL, Kutish GF, Ali A, Doster AR, Osorio FA. North American and European porcine reproductive and respiratory syndrome viruses differ in nonstructural protein coding regions. J Gen Virol. 1999;80(Pt 2):307–15.
Indik S, Schmoll F, Sipos W, Klein D. Genetic variability of PRRS virus in Austria: consequences for molecular diagnostics and viral quantification. Vet Microbiol. 2005;107:171–8.
Kim SH, Roh IS, Choi EJ, Lee C, Lee CH, Lee KH, Lee KK, Song YK, Lee OS, Park CK. A molecular analysis of European porcine reproductive and respiratory syndrome virus isolated in South Korea. Vet Microbiol. 2010;143:394–400.
Lee C, Kim H, Kang B, Yeom M, Han S, Moon H, Park S, Song D, Park B. Prevalence and phylogenetic analysis of the isolated type I porcine reproductive and respiratory syndrome virus from 2007 to 2008 in Korea. Virus Genes. 2010;40:225–30.
Thanawongnuwech R, Amonsin A, Tatsanakit A, Damrongwatanapokin S. Genetics and geographical variation of porcine reproductive and respiratory syndrome virus (PRRSV) in Thailand. Vet Microbiol. 2004;101:9–21.
Tun HM, Shi M, Wong CL, Ayudhya SN, Amonsin A, Thanawonguwech R, Leung FC. Genetic diversity and multiple introductions of porcine reproductive and respiratory syndrome viruses in Thailand. Virol J. 2011;8:164.
Christopher-Hennings J, Nelson E, Hines R, Nelson J, Swenson S, Zimmerman J, Chase C, Yaeger M, Benfield D. Persistence of porcine reproductive and respiratory syndrome virus in serum and semen of adult boars. J Vet Diagn Invest. 1995;7:456–64.
Kapur V, Elam MR, Pawlovich TM, Murtaugh MP. Genetic variation in porcine reproductive and respiratory syndrome virus isolates in the midwestern United States. J Gen Virol. 1996;77:1271–76.
Halbur PG, Paul PS, Meng XJ, Lum MA, Andrews JJ, Rathje JA. Comparative pathogenicity of nine US porcine reproductive and respiratory syndrome virus (PRRSV) isolates in a five-week-old cesarean-derived, colostrum-deprived pig model. J Vet Diagn Investig. 1996;8:11–20.
Meng XJ. Heterogeneity of porcine reproductive and respiratory syndrome virus: Implications for current vaccine efficacy and future vaccine development. Vet Microbiol. 2000;74:309–29.
OIE. Miscellaneous: “Swine high fever disease” in pigs in China (People’s Rep. of), Disease Information (Weekly info), 11 November 2005. 2006;18(45). http://web.oie.int/eng/info/hebdo/AIS_78.HTM#Sec15.
Youjun F, Zhao T, Nguyen T, Inui K, Ma Y, Nguyen TH, Nguyen VC, Liu D, Bui QA, To LT, Wang C, Tian K, Gao GF. Porcine Respiratory and Reproductive Syndrome Virus Variants, Vietnam and China, 2007. Emerg Infect Dis. 2008;14(11):1774–6.
Ni J, Yang S, Bounlom D, Yu X, Zhou Z, Song J, Khamphouth V, Vatthana T, Tian K. Emergence and pathogenicity of highly pathogenic Porcine reproductive and respiratory syndrome virus in Vientiane, Lao People’s Democratic Republic. J Vet Diagn Invest. 2012;24(2):349–54.
ProMED-mail. Porcine reprod. & resp. syndrome—Thailand. OIE; 2010. www.promedmail.org/direct.php?id=20101201.4320. Accessed 12 Oct 2010.
ProMED-mail. Porcine reprod. & resp. syndrome—Myanmar. Naypyidaw; 2011. 20110427.1307 http://www.promedmail.org/post/20110427.1307. Accessed 17 Nov 2011.
ProMED-mail. Undiagnosed disease, porcine—Cambodia; 2010. RFI 20100803.2618. www.promedmail.org/direct.php?id=20100803.2618. Accessed 3 Aug 2010.
ProMED-mail. Classical swine fever & PRRS, Philippines: (KA), susp. 2010. RFI 20100901.3123. https://www.promedmail.org/post/20100901.3123. Accessed 10 Feb 2011.
Nguyen T. PRRS Control in the region. OIE; 2013.
FAO. Porcine Reproductive and Respiratory Syndrome (PRRS); EMPRES Bulletin (2–2007). 2007. ftp://ftp.fao.org/docrep/fao/011/ai340e/ai340e00.pdf. Accessed 17 Nov 2010.
Too HL. In: A Guide to Pig Diseases in Malaysia. Faculty of Veterinary Medicine. Malaysia: Universiti Putra Malaysia; 1997. pp. 158–161.
Jasbir S, Kamaruddin MI, Latiffah H. Update on Porcine reproductive and respiratory syndrome (PRRS) seroprevalence in Malaysia. EMPRES Transboundary Animal Diseases Bulletin, FAO. 13th International Symposium for the World Association of Veterinary Laboratory Diagnosticians (WAVLD). 2008;30:38–9. ftp://ftp.fao.org/docrep/fao/010/i0059e/i0059e00.pdf.
Vania KTL, Ooi PT. Characterization of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) strains at selected farms in Malaysia. The 24th Veterinary Association Malausia Congress. 21-23th September 2012. Malaysia: Marriot Putrajaya; 2012.
Jaganathan S, Ooi PT, Phang LY, Zeenathul NA, Tuam SM, Ong LP, Kalaiwaney M, How SP, Tee CY, Lim HC, Choo PY, Lim BK. An update on the status of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) isolated in Malaysia. Ho Chi Minh: The 6th Asian Pig Veterinary Society Congress; 2013. September 23–25, 2013.
den Boon JA, Faaberg KS, Meulenberg JJ, Wassenaar AL, Plagemann PG, Gorbalenya AE, Snijder EJ. Processing and evolution of the N-terminal region of the arterivirus replicase ORF1a protein: identification of two papain-like cysteine proteases. J Virol. 1995;69:4500–5.
Han J, Burkhart KM, Vaughn EM, Roof MB, Faaberg KS. Replication and expression analysis of PRRSV defective RNA. In: The Nidoviruses. US: Springer; 2006. p. 445–8.
Han J, Liu G, Wang Y, Faaberg KS. Identification of nonessential regions of the nsp2 replicase protein of porcine reproductive and respiratory syndrome virus strain VR-2332 for replication in cell culture. J Virol. 2007;81:9878–90.
Allende R, Lewis TL, Lu Z, Rock DL, Kutish GF, Ali A, Doster AR, Osorio FA. North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J Gen Virol. 1999;80:307–15.
Nelsen CJ, Murtaugh MP, Faaberg KS. Porcine reproductive and respiratory syndrome virus comparison: Divergent evolution on two continents. J Virol. 1999;73:270–80.
Allende R, Kutish GF, Laegreid W, Lu Z, Lewis TL, Rock DL, Friesen J, Galeota JA, Doster AR, Osorio FA. Mutations in the genome of porcine reproductive and respiratory syndrome virus responsible for the attenuation phenotype. Arch Virol. 2000;145:1149–61.
Shen S, Kwang J, Liu W, Liu DX. Determination of the complete nucleotide sequence of a vaccine strain of porcine reproductive and respiratory syndrome virus and identification of the Nsp2 gene with a unique insertion. Arch Virol. 2000;145:871–83.
Fang Y, Kim DY, Ropp S, Steen P, Christopher-Hennings J, Nelson EA, Rowland RR. Heterogeneity in Nsp2 of European-like porcine reproductive and respiratory syndrome viruses isolated in the United States. Virus Res. 2004;100:229–35.
Fang Y, Rowland RR, Roof M, Lunney JK, Christopher-Hennings J, Nelson EA. A full-length cDNA infectious clone of North American type 1 porcine reproductive and respiratory syndrome virus: expression of green fluorescent protein in the Nsp2 region. J Virol. 2006;80:11447–55.
Gao ZQ, Guo X, Yang HC. Genomic characterization of two Chinese isolates of porcine respiratory and reproductive syndrome virus. Arch Virol. 2004;149:1341–51.
Ropp SL, Wees CE, Fang Y, Nelson EA, Rossow KD, Bien M, Arndt B, Preszler S, Steen P, Christopher-Hennings J, Collins JE, Benfield DA, Faaberg KS. Characterization of emerging European-like porcine reproductive and respiratory syndrome virus isolates in the United States. J Virol. 2004;78:3684–703.
Han J, Wang Y, Faaberg KS. Complete genome analysis of RFLP 184 isolates of porcine reproductive and respiratory syndrome virus. Virus Res. 2006;122:175–82.
Kim WI, Lee DS, Johnson W, Roof M, Cha SH, Yoon KJ. Effect of genotypic and biotypic differences among PRRS viruses on the serologic assessment of pigs for virus infection. Vet Microbiol. 2007;123:1–14.
Ran ZG, Chen XY, Guo X, Ge XN, Yoon KJ, Yang HC. Recovery of viable porcine reproductive and respiratory syndrome virus from an infectious clone containing a partial deletion within the Nsp2-encoding region. Arch Virol. 2008;153(5):899–907.
Metwally S, Mohamed F, Faaberg K, Burrage T, Prarat M, Moran K, Bracht A, Mayr G, Berninger M, Koster L, To TL, Nguyen VL, Reising M, Landgraf J, Cox L, Lubroth J, Carrillo C. Pathogenicity and Molecular Characterization of Emerging Porcine Reproductive and Respiratory Syndrome Virus in Vietnam in 2007. Transbound Emerg Dis. 2010;57:315–29.
Halbur PG, Paul PS, Frey ML, Landgraf J, Eernisse K, Meng XJ, Lum MA, Andrews JJ, Rathje JA. Comparison of the pathogenicity of two U.S. porcine reproductive and respiratory syndrome virus isolates with that of the Lelystad virus. Vet Pathol. 1995;34:648–60.
Halbur PG, Paul PS, Frey ML, Landgraf J, Eernisse K, Meng XJ, Andrews JJ, Lum MA, Rathje JA. Comparison of the antigen distribution of two U.S. porcine reproductive and respiratory syndrome virus isolates with that of the Lelystad virus. Vet Pathol. 1996;33:159–70.
Mengeling WL, Lager KM, Vorwald AC. Clinical consequences of exposing pregnant gilts to strains of porcine reproductive and respiratory syndrome (PRRS) virus isolated from field cases of “atypical” PRRS. Am J Vet Res. 1998;59:1540–4.
Opriessnig T, Halbur PG, Yoon KJ, Pogranichniy RM, Harmon KM, Evans R, Key KF, Pallares FJ, Thomas P, Meng XJ. Comparison of Molecular and Biological Characteristics of a Modified Live Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Vaccine (Ingelvac PRRS MLV), the Parent Strain of the Vaccine (ATCC VR2332), ATCC VR2385, and Two Recent Field Isolates of PRRSV. J Virol. 2002;76(23):11837–44.
Meng XJ, Paul PS, Halbur PG. Molecular cloning and nucleotide sequencing of the 3-terminal genomic RNA of porcine reproductive and respiratory syndrome virus. J Gen Virol. 1994;75:1795–801.
Meng XJ, Paul PS, Halbur PG, Lum MA. Phylogenic analyses of the putative M (ORF 6) and N (ORF 7) genes of porcine reproductive and respiratory syndrome virus (PRRSV): implication for the existence of two genotypes of PRRSV in the USA and Europe. Arch Virol. 1995;140:745–55.
Meng XJ, Paul PS, Halbur PG, Morozov I. Sequence comparison of open reading frames 2 to 5 of low and high virulence United States isolates of porcine reproductive and respiratory syndrome virus. J Gen Virol. 1995;76:3181–8.
Meng XJ, Paul PS, Halbur PG, Lum MA. Characterization of a high-virulence US isolate of porcine reproductive and respiratory syndrome virus in a continuous cell line, ATCC CRL11171. J Vet Diagn Investig. 1996;8:374–81.
Vu HLX, Ma F, Laegreid WW, Pattnaik AK, Steffen D, Doster AR, Osorio FA. A synthetic porcine reproductive and respiratory syndrome virus strain confers unprecedented levels of heterologous protection. J Virol. 2015;89:12070–83.
Pesch S. Etablierung einer Nachweismethode fur die zwei Genotypen von PRRSV und ein Beitrag zu seiner molekularen Epidemiologie. Doctoral Vet. Med. Leipzig: Thesis, Veterinary Faculty Universitat Leipzig; 2003.
Truyen U, Wilhelm S, Genzow M, Schagemann G. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): A Ring Test Performed in Germany to Assess RT-PCR Detection Methods. J Vet Med. 2006;53(2):68–74.
Mardassi H, Mounir S, Dea S. Identification of major differences in the nucleocapsid protein genes of a Quebec strain and European strains of porcine reproductive and respiratory syndrome virus. J Gen Virol. 1994;75:681–5.
Feng Y, Zhao T, Nguyen T, Inui K, Ma Y, Nguyen TH, Nguyen VC, Liu D, Bui QA, To LT, Wang C, Tian K, Gao GF. Porcine respiratory and reproductive syndrome virus variants, Vietnam and China, 2007. Emerg Infect Dis. 2008;14(11):1774–6.
Acknowledgements
The authors would like to thank Vet Food Agro Diagnostics (M) Sdn. Bhd for providing the samples for this study and Rhone Ma Malaysia Sdn. Bhd. for funding the research. The authors would also like to thank Prof. Dr. Henry Too and Dr. Francois Joisel for their advice & contribution.
Funding
The study was funded by Rhone Ma Malaysia Sdn. Bhd.
Availability of data and materials
All data were included in situ in the manuscript as tables and figures. Sequences derived from this study are publicly available on Genbank®. The phylogenetic data was deposited into TreeBase (www.treebase.org) and is publicly available at http://purl.org/phylo/treebase/phylows/study/TB2:S20336. The study ID on Treebase is S20336.
Authors’ contributions
SJ participated in the conceptual aspect of the work, conceived the research, performed the experiments and wrote the manuscript. TCY and LWH collected the samples, documented the clinical signs and assisted in the research design. OPT, PLY, ZNA, HSP, YLS, CPY, LBK provided consultation, research advise and coordination. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Organ samples were sent to the laboratory for diagnostic investigation. These diagnostic samples were collected under the supervision of institution veterinarians. The study was conducted following the guidelines as stated in the Code of Practice for Care and use of Animals for Scientific Purposes as stipulated by Universiti Putra Malaysia and complied with the current guidelines for the care and use of animals and was approved by the Animal Care and Use Committee (ACUC), Faculty of Veterinary Medicine, Universiti Putra Malaysia. There was no experimental research done on the animals. No animals were deliberately sacrificed or injured. Every effort was made to minimize any distress or unnecessary culling.
Disclaimer
This document is provided for scientific purposes only. Any reference to a brand or trademark herein is for informational purposes only and for providing specific information and is not intended for commercial purpose, to dilute the rights of the respective owner (s) of the brand (s) or trademark (s) and does not imply recommendation or endorsement by the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
King, S.J., Ooi, P.T., Phang, L.Y. et al. Phylogenetic characterization of genes encoding for viral envelope glycoprotein (ORF5) and nucleocapsid protein (ORF7) of porcine reproductive & respiratory syndrome virus found in Malaysia in 2013 and 2014.. BMC Vet Res 13, 3 (2016). https://doi.org/10.1186/s12917-016-0933-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-016-0933-z