
Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that is globally increasing in prevalence. The rise of ASD can be partially attributed to diagnostic expansion and advocacy efforts; however, the interplay between genetic predisposition and modern environmental exposures is likely driving a true increase in incidence. A range of evidence indicates that prenatal exposures are critical. Infection during pregnancy, gestational diabetes, and maternal obesity are established risk factors for ASD. Emerging areas of research include the effects of maternal use of selective serotonin reuptake inhibitors, antibiotics, and exposure to toxicants during pregnancy on brain development and subsequent ASD. The underlying pathways of these risk factors remain uncertain, with varying levels of evidence implicating immune dysregulation, mitochondrial dysfunction, oxidative stress, gut microbiome alterations, and hormonal disruptions. This narrative review assesses the evidence of contributing prenatal environmental factors for ASD and associated mechanisms as potential targets for novel prevention strategies.
Similar content being viewed by others


Background
Autism spectrum disorder (ASD) is an umbrella term for a heterogeneous group of neurodevelopmental disorders characterized by deficits in social communication, repetitive and stereotypical behaviors, and intellectual disabilities of varying levels [1]. ASD characteristics persist throughout life but are primarily diagnosed during early childhood when atypical developmental trajectories emerge [2]. A recent systematic review of 71 studies estimated the global median prevalence of ASD between 2012 and 2022 to be 10 per 1000 children and adults [3]. Males are approximately 4.2 times more likely to be affected than females [3], with differences in social communication, camouflaging, and restrictive and repetitive behaviors evident between the sexes [4].
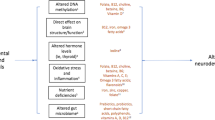
The etiology of ASD is multifactorial, shaped by inherited genetics, environmental contributions—particularly those associated with the maternal environment—and intricate gene–environment interactions [5, 6]. Epidemiological studies indicate that potential environmental prenatal factors contributing to ASD include exposure to infection during pregnancy, obesity, gestational diabetes mellitus (GDM), maternal selective serotonin reuptake inhibitor (SSRI) use, antibiotic use, and prenatal exposure to toxicants [7,8,9,10,11,12]. These prenatal factors may alter several pathways during critical developmental periods [13], particularly in genetically sensitive individuals [14]. The evidence relating to the potential pathways underlying the aforementioned prenatal factors is largely preclinical and implicates immune dysregulation, mitochondrial dysfunction, oxidative stress, gut microbiome alterations, and hormonal disruptions [15,16,17,18,19]. Understanding the complex etiology of ASD and underlying pathways may help identify early biomarkers and targets for primary prevention. While previous reviews have explored various genetic and prenatal factors associated with ASD risk [20, 21], here, we assess the current evidence for potentially modifiable prenatal environmental risk factors for ASD, as outlined in Fig. 1. We discuss potential mechanisms underlying each specific risk factor, particularly expanding on the role of the gut microbiome.

A schematic diagram summarizing prenatal environmental risk factors for autism spectrum disorder (ASD) and their potential mechanisms. From left to right, the left panel outlines genetic susceptibility and prenatal environmental risk factors such as infection during pregnancy, maternal obesity, diabetes, selective serotonin reuptake inhibitor (SSRI) and antibiotic use, and exposure to toxicants. The central panel details the underlying mechanisms for these risk factors, including inflammation, mitochondrial dysfunction, hormonal disruption, oxidative stress, and alterations in the gut microbiome that are associated with ASD development in offspring depicted in the panel on the right
Genetic predisposition
Many genes and gene variants likely contribute to ASD [22]. The high concordance of ASD in monozygotic twins, increased sibling recurrence, and familial aggregation of ASD suggest a strong genetic component, with heritability estimated to be approximately 50% [23,24,25,26]. The presence of autistic traits in genetically defined disorders, such as fragile X syndrome and Rett syndrome, supports the importance of genetic contributions to ASD more generally [27]. Sex differences in autism may arise due to differential genetic penetrance, sex-specific loci, or genetic mutations [28, 29]. Sex-specific phenotypic changes may begin via epigenetic interactions in utero through altered hormone exposure influencing endocrine function and neurodevelopment [30, 31]. A genetic predisposition to ASD may increase susceptibility to inflammation, mitochondrial dysfunction, oxidative stress, alterations in the gut microbiome, and hormone disruption, which may collectively underlie the connection between prenatal factors and ASD development [17, 32]. However, the clinical heterogeneity of ASD, including sexual dimorphism, behavioral phenotype distinctions, and individual developmental progression, makes it challenging to ascertain underlying genetic variations and pathophysiology [33].
Infection during pregnancy
Several studies have established an association between infections such as rubella [34, 35] and cytomegalovirus [36] during pregnancy and an increased likelihood of ASD in children [8, 37], consistent with the findings from preclinical studies investigating the role of maternal infection in ASD-like behavior in offspring [38]. A Danish population study found that viral infections including influenza, gastroenteritis, and other unspecified viral infections in the first trimester of pregnancy requiring hospitalization correlated with a 2.8-fold increase in the risk of ASD, while bacterial infections in the second trimester were associated with a 1.4-fold increase [39]. In the same cohort, exposure to only influenza during pregnancy was associated with a 2.3-fold increase in the risk of ASD, regardless of the trimester of exposure [40]. A meta-analysis of 36 studies revealed that any maternal infection or fever was associated with a 1.3-fold increase in ASD development [8]. Fever during pregnancy was associated with a 2.1-fold increase in the odds of ASD, independent of infection; however, these findings were attenuated in mothers who reported taking antipyretic medications to prevent or reduce fever, suggesting antipyretics may modify the relationship between infection and ASD [41]. A Danish population-based genome-wide association study found an association between the genetic profile of mothers with a history of any infection during pregnancy and subsequent ASD compared to the genetic profile of mothers with no history of infections during pregnancy [42]. While the reported genetic association between a history of maternal infection and ASD was relatively low, these maternal infection–based cases of ASD may have distinct genetic aetiologies. Validation of infection sites, types and timing during pregnancy poses a challenge, limiting the accurate evaluation of maternal infection’s role in ASD development. Further research is needed to distinguish the direct pathogenic effects of infection from the indirect effects of inflammation during pregnancy.
Potential mechanisms
Inflammation
Maternal immune activation (MIA), triggered by infection or inflammation during pregnancy, has been associated with a range of developmental difficulties in children, including increased risk of speech, language, and motor delays [43], increased behavioral and emotional problems [44] and altered connectivity in brain regions supporting working memory in children [45]. MIA elevates circulating and central proinflammatory cytokines in animal models, potentially impacting fetal immunity and neurodevelopment [46, 47]. Offspring of mice exposed to MIA during pregnancy exhibit heightened neuroinflammation and display ASD-like impaired inhibition and social interaction behaviors [48]. Maternal cytokines may cross the placenta, enter fetal circulation, and activate the fetal immune system [49, 50]. The placenta expresses all ten functional toll-like receptors, in humans and can therefore directly recognize microbial products, leading to increased placental inflammation and inflammatory cytokine release, affecting fetal brain development [51]. As such, administration of bacterial mimetic lipopolysaccharide (LPS) to pregnant mice on embryonic day 14 increases maternal circulating levels of interleukin (IL)-17, a T cell-derived proinflammatory cytokine, and subsequently leads to increased ASD-like impaired vocalization and social approach behaviors in their offspring [52]. Administration of IL-17-blocking antibodies prior to LPS prevented such behaviors [53]. In the poly(I:C) MIA mouse model, an increase in inflammatory cytokine concentration in maternal serum and amniotic fluid is associated with subsequent ASD-like deficits in inhibition control and exploratory and social behaviors in the offspring [54]. IL-6, a key proinflammatory cytokine, has been shown to play a critical role in promoting MIA-induced ASD-like deficits in inhibition control and social interaction in mice [48]. In addition to maternal IL-6 being able to cross the placenta and influence the fetus directly [55], there is also evidence that placental IL-6 signaling in response to MIA results in acute immune activation in the fetal brain and subsequent behavioral impairments [54]. In mice, blocking placental IL-6 signaling prevents MIA-induced immune activation in the fetal brain and subsequent behavioral abnormalities [54]. Elevated gene expression of IL-6 has also been observed in the brains of MIA-exposed mice and in the brain tissue of autistic individuals [56]. It is likely that concurrent risk factors or repeat infections are relevant to ASD susceptibility [57]. Adequate maternal levels of vitamins and antioxidants may enhance resilience to the effects of MIA [58], potentially influencing the effects of MIA on ASD development.
Mitochondrial function
Mitochondria are highly sensitive to immune and intrauterine changes and may mediate the relationship between infection-related inflammatory responses and ASD development [16]. A recent review identified 12 preclinical studies providing evidence for MIA-inducing ASD-like altered inhibitory and social behavior, sensorimotor coordination in offspring, and changes in brain mitochondrial morphology, density, and oxidative stress levels [59]. Exposure to LPS has been shown to increase reactive oxygen species (ROS) production [60] and lead to metabolic reprogramming from oxidative phosphorylation to glycolysis, particularly in microglia, the primary innate immune cells in the brain [61]. In addition to being a target of MIA, mitochondrial molecules, including adenosine triphosphate, mitochondrial DNA, and ROS, can also lead to inflammatory responses, directly influencing neuroinflammation [62]. Given the critical role of mitochondrial function in brain development, including neurogenesis, synaptogenesis, glial cell development and other processes with high bioenergetic requirements, MIA-induced disruption in mitochondrial bioenergetics during this critical period has major implications for neurodevelopment [59]. Preclinical models suggest the potential reversibility of MIA insults by redox modifiers such as N-acetylcysteine [63]. Human postmortem tissue studies have shown decreased expression of mitochondrial genes in regions involved in emotion and behavior regulation and motor function in autistic brains compared to non-autistic individuals [64]. Positron emission tomography (PET) scanning identified mitochondrial dysfunction in multiple brain regions in 23 adult autistic males compared to 24 age- and sex-matched nonautistic individuals [65]. Reduced mitochondrial function correlated with more severe social communication challenges in males, indicating a potential association between specific brain areas and core ASD characteristics [65]. The small sample size limits exploration of mitochondrial function using PET scanning and the study has yet to be repeated in females.
Gut microbiome
In preclinical studies, changes in the maternal gut microbiome during pregnancy mediate the effects of maternal infection on ASD development. The offspring of influenza A virus (IAV)-infected mice exhibited impaired mucosal immunity and altered immune cell profiles within gut-associated lymphoid tissue [66]. As IAV does not directly infect the fetus or placenta [67], this response may occur due to changes in the maternal immune system or potentially be mediated via the gut microbiota, which can influence early life immune development [66]. Offspring of the poly(I:C) MIA mouse model show ASD-like communicative, social, and stereotypic impairments alongside differences in the gut microbiome profile and increased intestinal permeability [18]. Oral administration of Bacteroides fragilis to MIA pups reduced gut permeability and ameliorated ASD-like anxiety, sensorimotor, repetitive, communicative, and social behaviors [18], suggesting that therapies targeting the gut microbiome may improve ASD-like behavioral symptoms. Mothers of children with ASD have more Proteobacteria, Alphaproteobacteria, Moraxellaceae, and Acinetobacter than mothers of children without ASD and that these microbiome profiles are associated with their children’s microbiome [68]. The role of the gut microbiome as a mediator between maternal infection and ASD development has yet to be explored in humans.
Infection during pregnancy induces inflammation, disrupting inflammatory pathways, and mitochondrial and gut microbiome function. While each pathway may play a distinct role in ASD development, there is likely a cumulative effect from multidirectional interactions between multiple pathways (Table 1). Variability in assessing acute and chronic inflammation highlights the necessity for standardized biomarkers and laboratory techniques to assess inflammatory responses.
Maternal obesity and GDM
Maternal obesity and GDM are both linked to an increased likelihood of ASD in children. Several epidemiological studies have shown that pre-pregnancy obesity increases the odds of ASD in children by 1.3–2.0-fold [7]. Additionally, excessive gestational weight gain on its own increases the odds for ASD in children by 1.1–1.6-fold [7]. A meta-analysis of 12 studies found that GDM was associated with a 1.5–1.7-fold increase in the risk for ASD in children compared to mothers without GDM [96]. Pre-existing type 2 diabetes is also associated with a 1.3-fold increase in the risk of ASD in children, and GDM is associated with a 1.4-fold increase when diagnosed at 26 weeks gestation or earlier [97]. No association between GDM and ASD development was detected when GDM was diagnosed after 26 weeks [97], indicating that the timing of GDM onset may be important.
Potential mechanisms
Inflammation
Both obesity and GDM are well-known inflammatory conditions and can lead to a dysregulated immune system, which may subsequently impair fetal neurodevelopment and increase ASD susceptibility [98]. Preclinical findings demonstrate that systemic inflammation induced by maternal obesity and GDM leads to an increased release of maternal proinflammatory cytokines that cross the placenta and alter the fetal development of neural pathways that regulate behavior [69]. Pregnant rats were administered streptozotocin, a diabetes-inducing agent, leading to elevated levels of inflammatory cytokines IL-1β and tumor necrosis factor (TNF)-α in the brain tissue of their offspring, accompanied by neurodevelopmental delays in behavioral tasks [73]. Exposure to a high-fat diet during pregnancy in rodents also increases local and systemic inflammation and the production of the proinflammatory cytokines, including IL-1β, IL-6, and TNF-α in maternal and offspring serum and brain, increasing atypical anxiety, cognitive and social behaviors in the offspring [70,71,72].
Hormonal disruption
Obesity and GDM may promote hormonal changes in women during pregnancy influencing maternal weight, glucose metabolism, and immune function [89, 97]. Consequently, obesity and GDM may disrupt the interaction between maternal metabolic hormones such as leptin and insulin [90, 99], leading to heightened secretion of inflammatory cytokines [100]. The dysregulation of metabolic hormones and cytokine secretion can be transmitted from mother to fetus, influencing the development of brain regions and neural circuitry associated with energy balance and behavior regulation [101]. Additionally, maternal and fetal metabolic hormone changes can shape the developing brain and may contribute to sexually dimorphic characteristics and behaviors [102, 103]. Mouse and human studies indicate that placental immune responses may occur in a sex-specific manner in response to maternal stressors, potentially mediated by hormonal disruptions and consequently influencing neurodevelopmental outcomes [104, 105]. However, whether placental immune activation has a causal role or reflects fetal brain immune activation in response to maternal obesity and diabetes has yet to be established, as has whether these responses reflect sex-specific ASD characteristics.
Gut microbiome
Obesity and GDM in women during pregnancy can alter the gut microbiota composition [80, 81] and lead to inflammation in the gut mucosa and impaired gut barrier integrity [72, 80]. An impaired gut barrier can result in the translocation of the mother’s gut microbiota and its by-products such as proinflammatory cytokines and LPS, into the placenta [84, 85]. In mice, depletion of the maternal gut microbiota inhibits placental growth and vascularisation, modulating metabolite circulation [82]. Supplementation with short-chain fatty acids (SCFAs) prevents the inhibition of placental growth and vascularisation, highlighting the crucial role of the maternal gut microbiome in early placental structure and function [82]. By contrast, the administration of neurotoxic levels of the SCFA propionic acid to pregnant mice induced ASD-like social, anxiety, and repetitive behaviors, which were alleviated by sodium butyrate, indicating gut-derived metabolites may have complex effects on ASD-like behavioral outcomes [83]. Neurotoxic levels of propionic acid also induced ASD-like behaviors in neonatal rats born to mothers with type 1 diabetes, and insulin therapy in the mothers mitigated ASD-like social and communication behaviors in offspring [106], highlighting the impact of hyperglycemia on offspring ASD and the potential benefits of insulin therapy during pregnancy.
Further investigation is needed to explore the relationship between obesity, GDM, and ASD, as well as potential underlying pathways, particularly in human cohorts. Discrepancies in epidemiological evidence may stem from variations in body mass index profiles and diabetes types considered. The co-occurrence of obesity and GDM, given their shared metabolic features and inflammatory profiles, complicates the assessment of their respective roles in ASD development, with the combined effect of maternal obesity and diabetes likely exerting a greater influence on ASD outcomes than either condition alone. Investigating the individual and combined contribution of obesity and GDM is crucial for identifying unique and shared underlying pathways. Prenatal dietary interventions targeting gut microbiota-derived inflammation could reduce the effect of maternal obesity and GDM on ASD development.
Maternal SSRI use
Evidence from epidemiological studies suggests that prenatal use of SSRI antidepressants is associated with a 1.5–4.5-fold increase in the risk for ASD in children, compared to children not exposed to SSRIs [11, 107,108,109]. This risk appears to be influenced at least to some extent by the trimester of pregnancy during which exposure to SSRIs occurred, with the highest odds of ASD development being for children of mothers with SSRI use during the first trimester [110, 111]. This timing of vulnerability to the effects of SSRIs coincides with the period of active organogenesis occurring between weeks three and eight of gestation, being also the critical period for ASD development in children of women exposed to potent teratogens, such as valproic acid and thalidomide [112, 113]. The association between prenatal SSRI use and ASD risk is attenuated when controlled for pre-existing psychiatric conditions [114], suggesting confounding by indication. Further, the significant association of preconception-only use of SSRIs, defined as occurring from 3 months prior to conception, and the risk of ASD in children, suggests that confounding by indication is likely involved [110]. Given the relatively short half-lives of SSRIs, except fluoxetine which can still be biologically active after five or more weeks of cessation [115], the mechanisms underlying the association between preconception exposure to SSRIs and ASD development are unclear. Additional population-based studies are required to improve the current understanding of risks versus benefits associated with SSRIs and other antidepressants used during pregnancy, particularly given the high risk of ASD development in children of mothers with psychiatric disorders [116].
Potential mechanisms
SSRI use during pregnancy may alter fetal serotonin system development, impair mitochondrial and gut microbiome function, and elevate oxidative stress [87, 93]. SSRIs act by inhibiting serotonin uptake centrally and peripherally [117]. During pregnancy, there is a substantial increase in peripheral serotonin production. In addition to gut enterochromaffin cells [118], the pancreas also contributes to increases in maternal serotonin levels during pregnancy [119], while placental production of serotonin contributes to the fetal brain serotonin concentration [120]. The serotonin system is critical for neurodevelopment, and its disruption has been implicated in ASD development, with elevated whole blood serotonin levels being consistently found in people with ASD [74]. High blood levels of serotonin in mothers exposed to SSRIs could therefore result in disrupted serotonin functioning in the fetal brain, leading to neurodevelopmental abnormalities [121]. Preclinical studies have shown that using SSRIs modulates mitochondrial function by affecting the activity of mitochondrial enzymatic complexes involved in the electron transport chain and the process of oxidative phosphorylation [78, 79]. While some studies have shown positive effects of certain SSRIs on the activities of individual enzymatic complexes or respiration analysis indicative of mitochondrial dysfunction, others have reported negative or divergent effects [79], with prenatal exposure potentially contributing to changes in neonatal energy metabolism [122]. It is, however, important to note that these conclusions are based on outcomes in animal studies, with almost no human data available. The gut microbiome is also essential for serotonin production, and gut microbes acting through short-chain fatty acids have been shown to modulate serotonin production in preclinical and in vitro human models [86]. Alterations in serotonin signaling observed in individuals with ASD following SSRI use may therefore be mediated by SSRI-induced changes in the gut microbiome [118, 123].
Epidemiological studies are needed to establish causal pathways between maternal SSRI use and ASD development. Isolating the direct impact of SSRIs is complex due to the interplay of depression, potential comorbidities, and lifestyle factors as well as the need to account for confounders like past maternal mental illness and timing of SSRI exposure. Studies of SSRI use may be influenced by indication bias, as the reasons for usage may affect the biological mechanisms underpinning ASD.
Maternal antibiotic use
Maternal antibiotic use during pregnancy may increase the likelihood of ASD, with 1.1–1.5-fold increases in the risk of ASD reported across several international cohorts after prenatal antibiotic exposure [12, 124, 125]. A prospective birth cohort showed that among women who received antibiotics during pregnancy, influenza in the second trimester was not associated with ASD development in children. For women who were not exposed to an antibiotic at any point during pregnancy, influenza in the second trimester was associated with a 4.0-fold increase in the odds of ASD, indicating that antibiotic use may modify the influence that maternal influenza has on ASD development [126]. Notably, adjusting for the type of antibiotic prescribed and using sibling-matched cohorts to account for unmeasured familial environmental and genetic confounders attenuated many of these findings. Studies on antibiotic use, like those on SSRIs, are often confounded by indication and include any exposure to antibiotics. Prospective cohort studies may help determine whether observed effects are due to antibiotics or underlying infections.
Potential mechanisms
Mouse models have demonstrated that antibiotic use can modify MIA’s effect on offspring neurodevelopmental outcomes, leading to behavioral abnormalities [15]. Antibiotic use can increase gut permeability [88], resulting in increased systemic translocation of the maternal gut microbiota and its by-products, such as LPS, into the placenta and fetal gut [88]. This, in turn, can impact fetal immunity or induce epigenetic changes by releasing SCFAs [88]. The administration of the broad-spectrum antibiotic vancomycin to pregnant poly(I:C) MIA mice altered the gut microbiota composition, resulting in reduced intestinal T helper 17 cell levels and decreased IL-17a in the maternal plasma, preventing reduced sociability, increased repetitive behaviors, and abnormal communication typically observed in the offspring of mice receiving MIA manipulation [75]. While these findings conflict with evidence that antibiotic use may contribute to ASD development, the presence of commensal bacteria that are sensitive to vancomycin may be necessary for the prevention of MIA-associated behavior in the offspring [75]. It is noteworthy that experimental antibiotic administration in rodents differs greatly from human usage in terms of types, dosages, treatment durations, and underlying conditions and reason for administration. Additionally, existing gut microbiome composition, immune response, and infection susceptibility vary due to species-specific physiology and future studies should consider these factors.
Maternal exposure to toxicants
Prenatal exposure to toxicants such as phthalates, air pollutants, and heavy metals may disrupt the endocrine system and increase inflammation, altering downstream neurodevelopment and increasing ASD likelihood [127]. Exposure to plasticizers such as phthalates measured in prenatal urine samples has been associated with higher ASD symptom scores in several epidemiological studies [10, 128, 129]. Prenatal air pollution exposure was also associated with a 2.2-3.6 increase in ASD risk in children in several large-scale epidemiological studies [130, 131], while a meta-analysis of 28 studies revealed that maternal exposure to air pollution was associated with a 1.4-fold increase in the risk of ASD in newborns [132]. A meta-analysis of two longitudinal cohort studies found that an increase in maternal concentrations of the heavy metals cadmium and cesium were both associated with a 1.8-fold increase in the risk of ASD [133]. A systematic review of 53 studies revealed higher lead and mercury concentrations in autistic individuals compared to non-autistic individuals [134]. However, significant geographical and metal detection heterogeneity among studies included in the meta-analysis highlights the need for future longitudinal studies with harmonized designs, measures, and analysis strategies. Evidence for an association between prenatal pesticide exposure and ASD is emerging but limited by variability in pesticide types [135,136,137], measurement methods, and exposure levels [138] between studies and there are few preclinical studies.
Potential mechanisms
The accumulation of persistent organic and heavy metal pollutants may act synergistically via alterations in hormonal pathways, inflammation, and oxidative stress, leading to neurotoxic effects during critical prenatal and early postnatal periods. Preclinically, stronger associations between maternal toxicant exposure and social impairment are often observed in males [139]. Stronger associations in males suggest a sex-specific interaction, potentially mediated by a hormone imbalance as maternal toxicant exposure can disrupt testosterone activity in the fetus [91]. By mimicking or interfering with hormone production or homeostasis, toxicants may influence sexually dimorphic aspects of neurodevelopment and subsequent social behaviors [139]. Further, bisphenol A exposure during pregnancy also increases male infants’ risk of ASD via downregulating methylation (silencing) of the brain aromatase gene, leading to decreased neuroestrogen production [92]. A large birth cohort study has demonstrated elevated prenatal di-(2-ethylhexyl) phthalate (DEHP) levels to be associated with increased ASD symptoms at 2 and 4 years, potentially mediated by a metabolic shift in pregnancy toward nonoxidative energy pathways [94]. Higher prenatal phthalate levels and a higher oxidative stress genetic score are associated with subsequent ASD, with oxidative stress-related single nucleotide polymorphisms (SNPs) modifying the prenatal phthalate and ASD association [10, 95]. Autistic children may metabolize toxicants differently [140], possibly due to epigenetic modifications that affect mechanisms underlying genetic susceptibility to toxicant exposure. Alteration in the genes that encode the proteins responsible for the breakdown of toxicants is one biologically plausible mechanism [141].
Diesel exhaust particles (DEPs), a primary toxic component of air pollution have been linked to ASD-like vocalization and social behavior deficits potentially mediated by dysregulated prenatal toll-like receptor (TLR)‑4 expression [77]. Prenatal exposure to DEPs in TLR-4 knockout mice induced neuroinflammation, delayed microglial maturation, and increased the levels of the inflammatory cytokine IL-1β, potentially linking DEP exposure to ASD development through immune dysregulation [76]. Heavy metals may also alter glutathione antioxidant capability, which has been found in individuals with ASD [142]; however, the relationship between maternal exposure to heavy metals and ASD development is not well understood in humans, and even less is known about the underlying mechanisms.
Research on the role of toxicants such as phthalates, air pollutants, and heavy metals in ASD development is still in its infancy and requires further investigation. To explore the underlying pathways, toxicant biomarkers are frequently measured in blood, urine, or hair. Assessing the accumulation of toxicants may benefit from using brain or tooth samples as they represent the tissue where toxicants are deposited [131]; however, obtaining these samples can be challenging, with brain tissue notably harder to collect than teeth.
Future directions
The last decade has seen major advances in identifying prenatal risk factors for ASD including exposure to infection, GDM, and obesity [7,8,9, 143], while the potential impacts of SSRI use, antibiotic use and exposure to toxicants are emerging fields of research [10,11,12]. Exploration of mechanisms underlying ASD such as immune dysregulation, gut microbiome alterations, hormonal disruption, mitochondrial dysfunction, and oxidative stress is predominantly preclinical. Transitioning preclinical findings into epidemiological studies is required to strengthen the evidence of pathways underpinning ASD and to identify potential targets for novel prevention strategies.
ASD is phenotypically and genetically heterogeneous, with the former creating diagnostic and classification challenges and the latter leading to a range of sensitivities to environmental risk factors, potentially masking relevant associations. ASD frequently co-occurs with other conditions and shares overlapping social, cognitive, and behavioral characteristics with ADHD, mood, and psychiatric disorders [144, 145]. Understanding how prenatal risk factors for ASD influence these shared characteristics may provide insights into related conditions. Improvement in the definition of ASD subgroups may reduce the diluting impact of case misclassification. Latent class analysis is a statistical method that can be used to identify ASD subgroups agnostically by analyzing the behavior and social traits of autistic individuals, to identify subgroups based on the severity of characteristics, co-occurring conditions, developmental trajectories, and potential responses to intervention strategies. Applying latent class analysis may increase our understanding of ASD heterogeneity and help identify biological pathways and treatment targets relevant to specific subgroups, thus enabling personalized management plans and improved outcomes for autistic individuals. The use of genetic susceptibility scores, like those evaluating the role of genetic predisposition to oxidative stress in ASD pathogenesis [95], may lead to an improved understanding of genetic susceptibility to environmental risk factors and their underlying biological processes [95]. Genetic susceptibility scores capturing the transcriptional activity of response pathways beyond oxidative stress may provide novel genetic markers of mechanisms associated with ASD development.
To address challenges in identifying causal pathways in epidemiological research, sufficiently large and detailed longitudinal birth cohort studies are needed to develop a comprehensive understanding of the impact of non-etiological factors and prenatal and early life risk factors on the developmental trajectory of ASD. Epidemiological studies must employ robust clinical phenotyping of the diverse spectrum of autistic traits and behaviors and integrate highly dimensioned multi-omic methodologies to offer valuable insights into the mechanisms underlying ASD. The exposome approach, which describes all an individual's environmental exposures throughout life, beginning with prenatal development [146], may contextualize multi-omic methods and enhance our understanding of gene–environment interactions on health outcomes, such as ASD development.
Epidemiological research must thoroughly consider potential confounding and mediating factors across pregnancy and early life. Complementing epidemiological findings with preclinical research is essential for testing causality and conducting more invasive investigations into the mechanisms underlying ASD development. Further research exploring the causation of ASD should examine maternal and early-life genetic and environmental contributors, focusing on identifying points of confluence in overlapping biological pathways to uncover predictive biomarkers and targets for early primary prevention.
Conclusions
ASD is a growing public health challenge, where the interplay between genetic predisposition and modern environmental exposures, notably the prenatal environment, plays a substantial role. Infection, obesity, GDM, SSRI use, antibiotics, and exposure to toxicants during pregnancy may all influence inflammation, hormonal balance, mitochondrial function, and the gut microbiome to disrupt subsequent neurodevelopment. Further, underlying pathways may interact, leading to cumulative effects on ASD risk. Understanding the interplay between genetics and the prenatal environment in ASD etiology and identifying modifiable points of confluence between multiple pathways, is crucial to developing novel and effective public health policies aimed at reducing prenatal exposure to risk factors in susceptible populations.
Availability of data and materials
Not applicable.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ASD:
-
Autism spectrum disorder
- DEPs:
-
Diesel exhaust particles
- DEHP:
-
Di-(2-ethylhexyl) phthalate
- GDM:
-
Gestational diabetes mellitus
- IAV:
-
Influenza A virus
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MIA:
-
Maternal immune activation
- PET:
-
Positron emission tomography
- SCFAs:
-
Short-chain fatty acids
- SNPs:
-
Single nucleotide polymorphisms
- SSRIs:
-
Selective serotonin reuptake inhibitors
- TNF:
-
Tumor necrosis factor
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. 2013.
Hyman SL, Levy SE, Myers SM, Kuo DZ, Apkon S, Davidson LF, et al. Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics. 2020;145(1):e20193447.
Zeidan J, Fombonne E, Scorah J, Ibrahim A, Durkin MS, Saxena S, et al. Global prevalence of autism: a systematic review update. Autism Res. 2022;15(5):778–90.
Ratto AB, Kenworthy L, Yerys BE, Bascom J, Wieckowski AT, White SW, et al. What about the girls? Sex-based differences in autistic traits and adaptive skills. J Autism Dev Disord. 2018;48(5):1698–711.
Hansen SN, Schendel DE, Parner ET. Explaining the Increase in the prevalence of autism spectrum disorders. JAMA Pediatr. 2015;169(1):56.
Frye RE, Cakir J, Rose S, Palmer RF, Austin C, Curtin P. Physiological mediators of prenatal environmental influences in autism spectrum disorder. BioEssays. 2021;43(9):2000307.
Tong L, Kalish BT. The impact of maternal obesity on childhood neurodevelopment. J Perinatol. 2021;41(5):928–39.
Tioleco N, Silberman AE, Stratigos K, Banerjee-Basu S, Spann MN, Whitaker AH, et al. Prenatal maternal infection and risk for autism in offspring: a meta-analysis. Autism Res. 2021;14(6):1296–316.
Rowland J, Wilson CA. The association between gestational diabetes and ASD and ADHD: a systematic review and meta-analysis. Sci Rep. 2021;11(1):5136.
Ponsonby A-L, Symeonides C, Saffery R, Mueller JF, O’Hely M, Sly PD, et al. Prenatal phthalate exposure, oxidative stress-related genetic vulnerability and early life neurodevelopment: a birth cohort study. Neurotoxicology. 2020;80:20–8.
Hagberg KW, Robijn AL, Jick SS. Maternal depression and antidepressant use during pregnancy and the risk of autism spectrum disorder in offspring. Clin Epidemiol. 2018;10:1599–612.
Hamad AF, Alessi-Severini S, Mahmud SM, Brownell M, Kuo IF. Prenatal antibiotics exposure and the risk of autism spectrum disorders: A population-based cohort study. PLoS One. 2019;14(8):e0221921.
Chaste P, Leboyer M. Autism risk factors: genes, environment, and gene-environment interactions. Dialogues Clin Neurosci. 2012;14(3):281–92.
Qiu S, Qiu Y, Li Y, Cong X. Genetics of autism spectrum disorder: an umbrella review of systematic reviews and meta-analyses. Transl Psychiatry. 2022;12(1):249.
Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry. 2017;81(5):391–401.
Frye RE, Cakir J, Rose S, Palmer RF, Austin C, Curtin P, et al. Mitochondria may mediate prenatal environmental influences in autism spectrum disorder. J Pers Med. 2021;11(3):218.
Mandic-Maravic V, Mitkovic-Voncina M, Pljesa-Ercegovac M, Savic-Radojevic A, Djordjevic M, Pekmezovic T, et al. Autism spectrum disorders and perinatal complications—Is oxidative stress the connection? Front Psychiatry. 2019;10:675.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–63.
Worsham W, Dalton S, Bilder DA. The prenatal hormone milieu in autism spectrum disorder. Front Psychiatry. 2021;12:655438.
Ornoy A, Weinstein-Fudim L, Ergaz Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod Toxicol. 2015;56:155–69.
Tordjman S, Somogyi E, Coulon N, Kermarrec S, Cohen D, Bronsard G, et al. Gene× Environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front Psych. 2014;5:53.
Schaaf CP, Betancur C, Yuen RKC, Parr JR, Skuse DH, Gallagher L, et al. A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat Rev Genet. 2020;21(6):367–76.
Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009;163(10):907.
Hansen SN, Schendel DE, Francis RW, Windham GC, Bresnahan M, Levine SZ, et al. Recurrence risk of autism in siblings and cousins: a multinational, population-based study. J Am Acad Child Adolesc Psychiatry. 2019;58(9):866–75.
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25(1):63–77.
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–9.
Quesnel-Vallières M, Weatheritt RJ, Cordes SP, Blencowe BJ. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat Rev Genet. 2019;20(1):51–63.
Goin-Kochel RP, Abbacchi A, Constantino JN, Autism Genetic Resource Exchange C. Lack of evidence for increased genetic loading for autism among families of affected females. Autism. 2007;11(3):279–86.
Szatmari P, Liu XQ, Goldberg J, Zwaigenbaum L, Paterson AD, Woodbury-Smith M, et al. Sex differences in repetitive stereotyped behaviors in autism: implications for genetic liability. Am J Med Genet B Neuropsychiatr Genet. 2012;159(1):5–12.
Baron-Cohen S, Lombardo MV, Auyeung B, Ashwin E, Chakrabarti B, Knickmeyer R. Why are autism spectrum conditions more prevalent in males? PLoS Biol. 2011;9(6):e1001081.
Napolitano A, Schiavi S, La Rosa P, Rossi-Espagnet MC, Petrillo S, Bottino F, et al. Sex differences in autism spectrum disorder: diagnostic, neurobiological, and behavioral features. Front Psychiatry. 2022;13:889636.
Cheroni C, Caporale N, Testa G. Autism spectrum disorder at the crossroad between genes and environment: contributions, convergences, and interactions in ASD developmental pathophysiology. Mol Autism. 2020;11(1):69.
Harris JC. The origin and natural history of autism spectrum disorders. Nat Neurosci. 2016;19(11):1390–1.
Chess S. Autism in children with congenital rubella. J Autism Child Schizophr. 1971;1(1):33–47.
Hutton J. Does rubella cause autism: a 2015 reappraisal? Front Hum Neurosci. 2016;10:25.
Yamashita Y, Fujimoto C, Nakajima E, Isagai T, Matsuishi T. Possible association between congenital cytomegalovirus infection and autistic disorder. J Autism Dev Disord. 2003;33(4):455–9.
Jiang H-Y, Xu L-L, Shao L, Xia R-M, Yu Z-H, Ling Z-X, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta-analysis. Brain Behav Immun. 2016;58:165–72.
Yin H, Wang Z, Liu J, Li Y, Liu L, Huang P, et al. Dysregulation of immune and metabolism pathways in maternal immune activation induces an increased risk of autism spectrum disorders. Life Sci. 2023;324:121734.
Atladóttir HÓ, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40(12):1423–30.
Atladottir HO, Henriksen TB, Schendel DE, Parner ET. Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics. 2012;130(6):e1447–54.
Zerbo O, Iosif A-M, Walker C, Ozonoff S, Hansen RL, Hertz-Picciotto I. Is Maternal Influenza or fever during pregnancy associated with autism or developmental delays? results from the CHARGE (CHildhood Autism Risks from Genetics and Environment) Study. J Autism Dev Disord. 2013;43(1):25–33.
Nudel R, Thompson WK, Børglum AD, Hougaard DM, Mortensen PB, Werge T, et al. Maternal pregnancy-related infections and autism spectrum disorder—the genetic perspective. Transl Psychiatry. 2022;12(1):334.
Girchenko P, Lahti-Pulkkinen M, Heinonen K, Reynolds RM, Laivuori H, Lipsanen J, et al. Persistently high levels of maternal antenatal inflammation are associated with and mediate the effect of prenatal environmental adversities on neurodevelopmental delay in the offspring. Biol Psychiat. 2020;87(10):898–907.
Patel S, Dale RC, Rose D, Heath B, Nordahl CW, Rogers S, et al. Maternal immune conditions are increased in males with autism spectrum disorders and are associated with behavioural and emotional but not cognitive co-morbidity. Transl Psychiatry. 2020;10(1):286.
Rudolph MD, Graham AM, Feczko E, Miranda-Dominguez O, Rasmussen JM, Nardos R, et al. Maternal IL-6 during pregnancy can be estimated from newborn brain connectivity and predicts future working memory in offspring. Nat Neurosci. 2018;21(5):765–72.
Tartaglione AM, Villani A, Ajmone-Cat MA, Minghetti L, Ricceri L, Pazienza V, et al. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl Psychiatry. 2022;12(1):384.
Bokobza C, Van Steenwinckel J, Mani S, Mezger V, Fleiss B, Gressens P. Neuroinflammation in preterm babies and autism spectrum disorders. Pediatr Res. 2019;85(2):155–65.
Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–702.
Wu W-L, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun. 2017;62:11–23.
Lim AI, McFadden T, Link VM, Han S-J, Karlsson R-M, Stacy A, et al. Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science. 2021;373(6558):eabf3002.
Koga K, Mor G. Toll-like receptors at the maternal–fetal interface in normal pregnancy and pregnancy disorders. Am J Reprod Immunol. 2010;63(6):587–600.
Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351(6276):933–9.
Yasumatsu K, Nagao JI, Arita-Morioka KI, Narita Y, Tasaki S, Toyoda K, et al. Bacterial-induced maternal interleukin-17A pathway promotes autistic-like behaviors in mouse offspring. Exp Anim. 2020;69(2):250–60.
Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25(4):604–15.
Dahlgren J, Samuelsson A-M, Jansson T, Holmäng A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60(2):147–51.
Wei H, Zou H, Sheikh AM, Malik M, Dobkin C, Brown WT, et al. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation. 2011;8(1):52.
Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science. 2016;353(6301):772–7.
Meyer U. Neurodevelopmental resilience and susceptibility to maternal immune activation. Trends Neurosci. 2019;42(11):793–806.
Gyllenhammer LE, Rasmussen JM, Bertele N, Halbing A, Entringer S, Wadhwa PD, et al. Maternal inflammation during pregnancy and offspring brain development: the role of mitochondria. Biol Psychiatry Cogn Neurosci Neuroimaging. 2022;7(5):498–509.
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457-70.e13.
Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia. 2019;67(6):1047–61.
M Wilkins H, H Swerdlow R. Relationships between mitochondria and neuroinflammation: implications for Alzheimer’s disease. Curr Top Med Chem. 2016;16(8):849–57.
Swanepoel T, Möller M, Harvey BH. N-acetyl cysteine reverses bio-behavioural changes induced by prenatal inflammation, adolescent methamphetamine exposure and combined challenges. Psychopharmacology. 2018;235(1):351–68.
Anitha A, Nakamura K, Thanseem I, Matsuzaki H, Miyachi T, Tsujii M, et al. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013;23(3):294–302.
Kato Y, Yokokura M, Iwabuchi T, Murayama C, Harada T, Goto T, et al. Lower availability of mitochondrial complex I in anterior cingulate cortex in autism: a positron emission tomography study. Am J Psychiatry. 2023;180(4):277–84.
Liong S, Miles MA, Mohsenipour M, Liong F, Hill-Yardin EL, Selemidis S. Influenza A virus infection during pregnancy causes immunological changes in gut-associated lymphoid tissues of offspring mice. Am J Physiol Gastrointest Liver Physiol. 2023;325(3):G230–8.
Liong S, Oseghale O, To EE, Brassington K, Erlich JR, Luong R, et al. Influenza A virus causes maternal and fetal pathology via innate and adaptive vascular inflammation in mice. Proc Natl Acad Sci. 2020;117(40):24964–73.
Li N, Yang J, Zhang J, Liang C, Wang Y, Chen B, et al. Correlation of gut microbiome between ASD children and mothers and potential biomarkers for risk assessment. Genomics Proteomics Bioinformatics. 2019;17(1):26–38.
Han VX, Patel S, Jones HF, Nielsen TC, Mohammad SS, Hofer MJ, et al. Maternal acute and chronic inflammation in pregnancy is associated with common neurodevelopmental disorders: a systematic review. Transl Psychiatry. 2021;11(1):71.
Bordeleau M, Lacabanne C, de Fernández Cossío L, Vernoux N, Savage JC, González-Ibáñez F, et al. Microglial and peripheral immune priming is partially sexually dimorphic in adolescent mouse offspring exposed to maternal high-fat diet. J Neuroinflamm. 2020;17:1–28.
Xavier S, Soch A, Younesi S, Malik S, Spencer SJ, Sominsky L. Maternal diet before and during pregnancy modulates microglial activation and neurogenesis in the postpartum rat brain. Brain Behav Immun. 2021;98:185–97.
Bilbo SD, Tsang V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 2010;24(6):2104–15.
Piazza FV, Segabinazi E, De Meireles ALF, Mega F, Spindler CDF, Augustin OA, et al. Severe uncontrolled maternal hyperglycemia induces microsomia and neurodevelopment delay accompanied by apoptosis, cellular survival, and neuroinflammatory deregulation in rat offspring hippocampus. Cell Mol Neurobiol. 2019;39(3):401–14.
Muller CL, Anacker AMJ, Veenstra-Vanderweele J. The serotonin system in autism spectrum disorder: from biomarker to animal models. Neuroscience. 2016;321:24–41.
Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549(7673):528–32.
Bolton JL, Marinero S, Hassanzadeh T, Natesan D, Le D, Belliveau C, et al. Gestational exposure to air pollution alters cortical volume, microglial morphology, and microglia-neuron interactions in a sex-specific manner. Front Synaptic Neurosci. 2017;9:10.
Xiao L, Feng J, Zhang W, Pan J, Wang M, Zhang C, et al. Autism-like behavior of murine offspring induced by prenatal exposure to progestin is associated with gastrointestinal dysfunction due to claudin-1 suppression. Febs j. 2023;290(13):3369–82.
de Oliveira MR. Fluoxetine and the mitochondria: a review of the toxicological aspects. Toxicol Lett. 2016;258:185–91.
Emmerzaal TL, Nijkamp G, Veldic M, Rahman S, Andreazza AC, Morava E, et al. Effect of neuropsychiatric medications on mitochondrial function: for better or for worse. Neurosci Biobehav Rev. 2021;127:555–71.
Houttu N, Mokkala K, Laitinen K. Overweight and obesity status in pregnant women are related to intestinal microbiota and serum metabolic and inflammatory profiles. Clin Nutr. 2018;37(6 Pt A):1955–66.
Kuang Y-S, Lu J-H, Li S-H, Li J-H, Yuan M-Y, He J-R, et al. Connections between the human gut microbiome and gestational diabetes mellitus. GigaScience. 2017;6(8):1–12.
Pronovost GN, Yu KB, Elena J, Telang SS, Chen AS, Vuong HE, et al. The maternal microbiome promotes placental development in mice. Sci Adv. 2023;9(40):eadk1887.
Kratsman N, Getselter D, Elliott E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology. 2016;102:136–45.
Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Backhed HK, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150(3):470–80.
Nuriel-Ohayon M, Neuman H, Koren O. Microbial changes during pregnancy, birth, and infancy. Front Microbiol. 2016;7:1031.
Reigstad CS, Salmonson CE, Rainey JF III, Szurszewski JH, Linden DR, Sonnenburg JL, et al. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015;29(4):1395.
Rodnyy AY, Kondaurova EM, Tsybko AS, Popova NK, Kudlay DA, Naumenko VS. The brain serotonin system in autism. Rev Neurosci. 2023;35(1):1–20. https://doi.org/10.1515/revneuro-2023-0055.
Chen CM, Chou HC, Yang YCSH. Maternal antibiotic treatment disrupts the intestinal microbiota and intestinal development in neonatal mice. Front Microbiol. 2021;12:684233.
Agrawal R, Agrawal A, Jacson MJ. Maternal obesity and autism spectrum disorders in offspring. Indian J Child Health. 2022;9(10):178–82.
Qiu C, Williams MA, Vadachkoria S, Frederick IO, Luthy DA. Increased maternal plasma leptin in early pregnancy and risk of gestational diabetes mellitus. Obstet Gynecol. 2004;103(3):519–25.
Braun JM, Kalkbrenner AE, Just AC, Yolton K, Calafat AM, Sjödin A, et al. Gestational exposure to endocrine-disrupting chemicals and reciprocal social, repetitive, and stereotypic behaviors in 4-and 5-year-old children: the HOME study. Environ Health Perspect. 2014;122(5):513–20.
Symeonides C, Vacy K, Thomson S, Tanner S, Chua HK, Dixit S, et al. Male autism spectrum disorder is linked to brain aromatase disruption by prenatal BPA in multimodal investigations and 10HDA ameliorates the related mouse phenotype. Nat Commun. 2024;15(1):6367.
Bhat RS, Alonazi M, Al-Daihan S, El-Ansary A. Prenatal SSRI exposure increases the risk of autism in rodents via aggravated oxidative stress and neurochemical changes in the brain. Metabolites. 2023;13(2):310.
Thomson S, Drummond K, O’Hely M, Symeonides C, Chandran C, Mansell T, et al. Increased maternal non-oxidative energy metabolism mediates association between prenatal di-(2-ethylhexyl) phthalate (DEHP) exposure and offspring autism spectrum disorder symptoms in early life: A birth cohort study. Environ Int. 2023;171:107678.
Tanner S, Thomson S, Drummond K, O’Hely M, Symeonides C, Mansell T, et al. A pathway-based genetic score for oxidative stress: an indicator of host vulnerability to phthalate-associated adverse neurodevelopment. Antioxidants. 2022;11(4):659.
Xu G, Jing J, Bowers K, Liu B, Bao W. Maternal diabetes and the risk of autism spectrum disorders in the offspring: a systematic review and meta-analysis. J Autism Dev Disord. 2014;44(4):766–75.
Xiang AH, Wang X, Martinez MP, Walthall JC, Curry ES, Page K, et al. Association of maternal diabetes with autism in offspring. JAMA. 2015;313(14):1425–34.
Kong L, Chen X, Gissler M, Lavebratt C. Relationship of prenatal maternal obesity and diabetes to offspring neurodevelopmental and psychiatric disorders: a narrative review. Int J Obes. 2020;44(10):1981–2000.
Ye W, Luo C, Huang J, Li C, Liu Z, Liu F. Gestational diabetes mellitus and adverse pregnancy outcomes: systematic review and meta-analysis. BMJ. 2022;377:e067946.
Khambule L, George JA. The role of inflammation in the development of GDM and the use of markers of inflammation in GDM screening. In: Guest, P. (eds) reviews on biomarker studies of metabolic and metabolism-related disorders. Advances in experimental medicine and biology, vol 1134. Cham: Springer; 2019. https://doi.org/10.1007/978-3-030-12668-1_12.
Sotgiu S, Manca S, Gagliano A, Minutolo A, Melis MC, Pisuttu G, et al. Immune regulation of neurodevelopment at the mother–foetus interface: the case of autism. Clin Transl Immunol. 2020;9(11):e1211.
Berenbaum SA, Beltz AM. How early hormones shape gender development. Curr Opin Behav Sci. 2016;7:53–60.
McCarthy MM. Multifaceted origins of sex differences in the brain. Philos Trans R Soc B Biol Sci. 2016;371(1688):20150106.
Behura SK, Kelleher AM, Spencer TE. Evidence for functional interactions between the placenta and brain in pregnant mice. FASEB J. 2019;33(3):4261.
Bale TL. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin Neurosci. 2016;18(4):459–64.
Aljumaiah MM, Alonazi MA, Al-Dbass AM, Almnaizel AT, Alahmed M, Soliman DA, et al. Association of maternal diabetes and autism spectrum disorders in offspring: a study in a rodent model of autism. J Mol Neurosci. 2022;72(2):349–58.
Harrington RA, Lee L-C, Crum RM, Zimmerman AW, Hertz-Picciotto I. Prenatal SSRI use and offspring with autism spectrum disorder or developmental delay. Pediatrics. 2014;133(5):e1241–8.
Sørensen MJ, Grønborg TK, Christensen J, Parner ET, Vestergaard M, Schendel D, et al. Antidepressant exposure in pregnancy and risk of autism spectrum disorders. Clin Epidemiol. 2013;5(null):449–59.
Malm H, Brown AS, Gissler M, Gyllenberg D, Hinkka-Yli-Salomäki S, McKeague IW, et al. Gestational exposure to selective serotonin reuptake inhibitors and offspring psychiatric disorders: a national register-based study. J Am Acad Child Adolesc Psychiatry. 2016;55(5):359–66.
Kaplan YC, Keskin-Arslan E, Acar S, Sozmen K. Prenatal selective serotonin reuptake inhibitor use and the risk of autism spectrum disorder in children: a systematic review and meta-analysis. Reprod Toxicol. 2016;66:31–43.
Mezzacappa A, Lasica P-A, Gianfagna F, Cazas O, Hardy P, Falissard B, et al. Risk for autism spectrum disorders according to period of prenatal antidepressant exposure: a systematic review and meta-analysis. JAMA Pediatr. 2017;171(6):555–63.
Ornoy A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod Toxicol. 2009;28(1):1–10.
Vargesson N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today. 2015;105(2):140–56.
Kobayashi T, Matsuyama T, Takeuchi M, Ito S. Autism spectrum disorder and prenatal exposure to selective serotonin reuptake inhibitors: a systematic review and meta-analysis. Reprod Toxicol. 2016;65:170–8.
Keks N, Hope J, Keogh S. Switching and stopping antidepressants. Aust Prescr. 2016;39(3):76.
Yin W, Pulakka A, Reichenberg A, Kolevzon A, Ludvigsson JF, Risnes K, et al. Association between parental psychiatric disorders and risk of offspring autism spectrum disorder: a Swedish and Finnish population-based cohort study. Lancet Regional Health Eur. 2024;40:100902.
Vaswani M, Linda FK, Ramesh S. Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(1):85–102.
Gershon MD. 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes. 2013;20(1):14.
Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16(7):804–8.
Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, et al. A transient placental source of serotonin for the fetal forebrain. Nature. 2011;472(7343):347–50.
Sujan AC, Öberg AS, Quinn PD, D’Onofrio BM. Annual Research Review: Maternal antidepressant use during pregnancy and offspring neurodevelopmental problems - a critical review and recommendations for future research. J Child Psychol Psychiatry. 2019;60(4):356–76.
da LD Barros M, Manhaes-de-castro R, Alves DT, Quevedo OG, Toscano AE, Bonnin A, et al. Long term effects of neonatal exposure to fluoxetine on energy balance: a systematic review of experimental studies. Eur J Pharmacol. 2018;833:298–306.
De Angelis M, Piccolo M, Vannini L, Siragusa S, De Giacomo A, Serrazzanetti DI, et al. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One. 2013;8(10):e76993.
Lee E, Cho J, Kim KY. The association between autism spectrum disorder and pre- and postnatal antibiotic exposure in childhood—A systematic review with meta-analysis. Int J Environ Res Public Health. 2019;16(20):4042.
Wimberley T, Agerbo E, Pedersen CB, Dalsgaard S, Horsdal HT, Mortensen PB, et al. Otitis media, antibiotics, and risk of autism spectrum disorder. Autism Res. 2018;11(10):1432–40.
Holingue C, Brucato M, Ladd-Acosta C, Hong X, Volk H, Mueller NT, et al. Interaction between maternal immune activation and antibiotic use during pregnancy and child risk of autism spectrum disorder. Autism Res. 2020;13(12):2230–41.
Moosa A, Shu H, Sarachana T, Hu VW. Are endocrine disrupting compounds environmental risk factors for autism spectrum disorder? Horm Behav. 2018;101:13–21.
Haggerty DK, Strakovsky RS, Talge NM, Carignan CC, Glazier-Essalmi AN, Ingersoll BR, et al. Prenatal phthalate exposures and autism spectrum disorder symptoms in low-risk children. Neurotoxicol Teratol. 2021;83:106947.
Oulhote Y, Lanphear B, Braun JM, Webster GM, Arbuckle TE, Etzel T, et al. Gestational exposures to phthalates and folic acid, and autistic traits in Canadian children. Environ Health Perspect. 2020;128(2):027004.
Volk HE, Hertz-Picciotto I, Delwiche L, Lurmann F, McConnell R. Residential proximity to freeways and autism in the CHARGE study. Environ Health Perspect. 2011;119(6):873–7.
Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry. 2014;4(2):e360.
Dutheil F, Comptour A, Morlon R, Mermillod M, Pereira B, Baker JS, et al. Autism spectrum disorder and air pollution: a systematic review and meta-analysis. Environ Pollut. 2021;278:116856.
Dou JF, Schmidt RJ, Volk HE, Nitta MM, Feinberg JI, Newschaffer CJ, Croen LA, Hertz-Picciotto I, Fallin MD, Bakulski KM. Exposure to heavy metals in utero and autism spectrum disorder at age 3: a meta-analysis of two longitudinal cohorts of siblings of children with autism. Environ Health. 2024;23(1):62. https://doi.org/10.1186/s12940-024-01101-2.
Ding M, Shi S, Qie S, Li J, Xi X. Association between heavy metals exposure (cadmium, lead, arsenic, mercury) and child autistic disorder: a systematic review and meta-analysis. Front Pediatr. 2023;11:1169733.
Lyall K, Croen LA, Sjödin A, Yoshida CK, Zerbo O, Kharrazi M, et al. Polychlorinated biphenyl and organochlorine pesticide concentrations in maternal mid-pregnancy serum samples: association with autism spectrum disorder and intellectual disability. Environ Health Perspect. 2017;125(3):474–80.
Schmidt RJ, Kogan V, Shelton JF, Delwiche L, Hansen RL, Ozonoff S, et al. Combined prenatal pesticide exposure and folic acid intake in relation to autism spectrum disorder. Environ Health Perspect. 2017;125(9):097007.
von Ehrenstein OS, Ling C, Cui X, Cockburn M, Park AS, Yu F, et al. Prenatal and infant exposure to ambient pesticides and autism spectrum disorder in children: population based case-control study. Bmj. 2019;364:l962.
Román P, Ruiz-González C, Rueda-Ruzafa L, Cardona D, Requena M, Alarcón R. Exposure to environmental pesticides and the risk of autism spectrum disorders: a population-based case-control study. Medicina. 2024;60(3):479.
Goodman CV, Green R, Dacosta A, Flora D, Lanphear B, Till C. Sex difference of pre- and post-natal exposure to six developmental neurotoxicants on intellectual abilities: a systematic review and meta-analysis of human studies. Environ Health. 2023;22(1):80.
Stamova B, Green PG, Tian Y, Hertz-Picciotto I, Pessah IN, Hansen R, et al. Correlations between gene expression and mercury levels in blood of boys with and without autism. Neurotox Res. 2011;19:31–48.
Ijomone OM, Olung NF, Akingbade GT, Okoh COA, Aschner M. Environmental influence on neurodevelopmental disorders: potential association of heavy metal exposure and autism. J Trace Elem Med Biol. 2020;62:126638.
Kern JK, Geier DA, Adams JB, Garver CR, Audhya T, Geier MR. A clinical trial of glutathione supplementation in autism spectrum disorders. Med Sci Monit. 2011;17(12):CR677.
Sandin S, Schendel D, Magnusson P, Hultman C, Surén P, Susser E, et al. Autism risk associated with parental age and with increasing difference in age between the parents. Mol Psychiatry. 2016;21(5):693–700.
Lugo Marín J, Alviani Rodríguez-Franco M, Mahtani Chugani V, MagánMaganto M, DíezVilloria E, Canal BR. Prevalence of schizophrenia spectrum disorders in average-IQ adults with autism spectrum disorders: a meta-analysis. J Autism Dev Disord. 2018;48(1):239–50.
Mutluer T, Aslan Genç H, Özcan Morey A, YapiciEser H, Ertinmaz B, Can M, et al. Population-based psychiatric comorbidity in children and adolescents with autism spectrum disorder: a meta-analysis. Front Psychiatry. 2022;13:856208.
Wild CP. The exposome: from concept to utility. Int J Epidemiol. 2012;41(1):24–32.
Acknowledgements
We acknowledge the lived experience of neurodivergent people and their contribution to humanity. We are aware that some individuals may prefer identity-first language while others may prefer person-first language; we use both in this paper. Figure 1 was created with BioRender.com.
Funding
MB is supported by a NHMRC Leadership 3 Investigator grant (GNT2017131). Samantha Dawson was supported by an Alfred Deakin Postdoctoral Research Fellowship and is currently supported by a Medical Research Future Fund grant (MRFF 2025947). No other funding directly contributed to this manuscript.
Author information
Authors and Affiliations
Contributions
CL and SD designed and conceived this manuscript. CL wrote the manuscript with major contributions from SD. LS made substantial contributions regarding maternal SSRI use. LS, MOH, MB and PV critically revised the manuscript and contributed to the final draft. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Love, C., Sominsky, L., O’Hely, M. et al. Prenatal environmental risk factors for autism spectrum disorder and their potential mechanisms. BMC Med 22, 393 (2024). https://doi.org/10.1186/s12916-024-03617-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-024-03617-3