
Abstract
Background
In this study, we investigated the anti-obesity and anti-hyperlipidemic mechanisms of lambertianic acid (LA) isolated from Pinus koraiensis leaves and the ethanol extract of Pinus koraiensis leaves (EPK), both in vitro and in vivo.
Methods
Differentiated 3T3L-1 cells were treated with EPK (25 or 50 μg/mL) or LA (200 μM) and analyzed by western blotting or RT-PCR. In vitro, lipid accumulation of adipocytes was observed using Oil-Red-O staining and triglyceride analysis. The contribution of AMPK to anti-obesity activity was assessed by siRNA-mediated AMPK knockdown. After AMPK silencing, expression of AMPK was observed by western blotting. To confirm the in vitro activity, an animal study was conducted by administering a normal diet, HFD, and EPK for 6 weeks. Obesity-related physiological parameters and protein levels were measured.
Results
LA induced the expression of p-AMPK and inhibited PPARγ, C/EBP α, adiponectin, FAS, SREBP-1, and HMGCR expression. EPK containing LA significantly decreased lipid accumulation and triglyceride levels in the differentiated 3 T3-L1 cells. EPK treatment suppressed the expression of adipogenic transcription factors, FABP, GPDH, and cholesterol-synthesis-related factors in the differentiated 3 T3-L1 cells. EPK increased the expression of p-AMPK. The effects of EPK were reversed on inhibiting AMPK by using AMPK siRNA and compound C. In vivo analysis showed that body weight gain, serum triglyceride, total cholesterol, LDL cholesterol and AI value in the EPK treatment group were lower than those in the HFD control group. EPK induced the expression of p-AMPK and inhibited PPARγ in liver and adipose tissue.
Conclusions
Overall, the results suggest that EPK containing LA exerts significant anti-obesity and cholesterol-lowering effects by activating AMPK.
Similar content being viewed by others


Background
Obesity is a complex multifactorial chronic disease characterized by excess body fat and is associated with concurrent diseases that reduce life expectancy, including cardiovascular disease, stroke, hyperlipidemia, fatty liver, and diabetes [1, 2]. Obesity caused by hypertrophy of adipose tissue as well as adipose tissue hyperplasia triggers the differentiation of preadipocytes into adipocytes [3]. Adipocyte differentiation is regulated by crucial transcription factors such as peroxisome proliferator-activated receptor-γ (PPARγ) and CCAAT/enhancer-binding proteins α (C/EBPα). These transcription factors control the expression of many adipogenic proteins [4–7]. Several studies have reported that Sterol regulatory element-binding protein 1 (SREBP-1) is a transcription factor that regulates adipogenesis, cellular cholesterol, and cholesterol synthesis proteins, such as 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), in 3 T3-L1 adipocytes [8–10]. Adenosine monophosphate-activated protein kinase (AMPK) is a key enzyme in energy metabolism and is involved in regulation of glucose levels and lipid uptake. AMPK is expressed in a number of tissues, including adipose tissue, the liver, skeletal muscle, the heart, pancreatic beta cells, and brain cells [11, 12]. AMPK is phosphorylated and inactivates metabolic enzymes involved in fatty acid and cholesterol syntheses [13–15]. AMPK also provides an upstream signal of PPARγ/CEBPα and suppresses differentiation of preadipocytes into adipocytes [16–18]. Activation of AMPK decreases cellular cholesterol and fatty acids, such as SREBP-1 and HMGCR [19, 20]. Recently, the identification of a natural compound that can exert anti-obesity effects with fewer side effects than currently available prescription medications is attracting attention [4]. One such compound is P. koraiensis (Korean nut pine), which is native to Korea, Japan, China, and Eastern Russia. The main chemicals in essential oil from P. koraiensis leaves (EOPK) are camphene, D-limonene, borneol, α-pinene, 3-carene, 4-carene, β-phellandrene, and fencyl [21]. Our previous research has shown that EOPK has anti-hyperlipidemic [21], anti-diabetic [22], anti-obesity [23] and anti-cancer effects [24]. P. koraiensis seed oil has been investigated to inhibition of lipid metabolism in rats and mice [25, 26]. However, the biological and biochemical effects of the ethanol extract of P. koraiensis (EPK) and its main compounds have not yet been proven. The EPK is easier to extract than EOPK. In addition, EPKs are convenient and easy to use. The purpose of this study is to investigate the anti-adipogenic effect of EPK on 3T3L-1 cells and the anti-obesity activity of EPK on high fat diet (HFD)-fed rats.
Methods
Plant materials
The leaves of P. koraiensis were used same materials with our previous study and the details were described in our previous published study [24].
Preparation of EPK
The EPK was prepared using the hydrodistillation method with dried and pulverized P. koraiensis leaves. In order to increase the extraction efficiency, P. koraiensis leaves and young stems under 1 cm in diameter were cut into 2–3 cm sections. P. koraiensis leaves were obtained from the agricultural corporation Beaksongyounlim (Gangwondo, Korea) and authenticated by the Department of Oriental Medicine Biotechnology at Kyung Hee University. Dried and pulverized leaves (1 kg) were immersed in 50 % ethanol (10 L) and distilled. The reflux distillation was continued for 10 h at 45 °C and was repeated twice. The EPK was prepared under reduced pressure to obtain 30 Brix.
Extraction and isolation of LA
The EPK was partitioned with EtOAc / distilled water (1:1). The water layer was suspended and partitioned with n-butanol / distilled water. Coulum chromatography of the EtOAc fraction showing anti-hyperlipidemic activity, over silica gel using an n-hexan-EtOAc-chloroform-MeOH mixture with increasing polarity, yielded 15 fractions. 15 fractions were confirmed with thin layer chromatography. Among these fractions, fr. 6 showed distinct and vivid red-purple color by TLC. Also the fr. 6 showed the most potent anti-hyperlipidemic aciticity. LA was obtained by an additional purification step from fr. 6. The structure of LA was identified by 1H-nuclear magnetic resonance (NMR) and 13C-nuclear magnetic resonance (NMR). (Additional file 1: Figure S1)
Cell culture assay
3T3L-1 preadipocytes were purchased from Korean Cell Line Bank (KCLB). The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4500 mg/L D-glucose, 10 % fetal bovine serum (FBS), 2 μM L-glutamine and penicillin/streptomycin (WelGene, Daegu, South Korea) in a humidified atmosphere of 5 % CO2 at 37 °C.
Cytotoxicity assay
Cytotoxicity of EPK was evaluated with the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) (Sigma Aldrich, St Louis, MO) assay. The cells were seeded at a density of 1 × 104cells per well in a 96-well plate, cultured for 24 h, and then treated with various concentrations of EPK. After 24 h incubation, 50 μL of MTT solution (1 mg/mL) was added to each well and incubated for 2 h at 37 °C in darkness. The viable cell number was correlated with the production of formazan, which was dissolved with dimethyl sulfoxide (DMSO), and optical density (O.D.) was measured with a microplate reader (Sunrise, TECAN, Mannedorf, Switzerland) at 570 nm. Cell viability was calculated by the following equation: Cell viability (%) = [O.D.(EPK)-O.D.(blank)]/[O.D(control)-O.D.(blank)] × 100.
Differentiation induction and Oil-Red-O staining
The preadipocyte 3 T3-L1 cells were plated on 6-well plates on day 0 and incubated until confluency was achieved. For adipocyte differentiation, the confluent cells were treated with 1 μM dexamethasone, 1 μg/mL insulin, and 0.5 mM IBMX for 2 days, and the medium was replaced by fresh normal medium containing only 1 μg/mL insulin for 2 days. On day 2, the differentiated adipocyte cells were cultured in the presence or absence of EPK (25 or 50 μg/mL) for 6 days. The medium was changed every 2 days. The cells were fixed with 2 % paraformaldehyde, washed twice with PBS, and finally stained with Oil-Red-O. The cellular lipid retained Oil-Red-O in isopropanol and adipocyte expression was estimated by measuring O.D. with a microplate reader (Sunrise, TECAN, Mannedorf, Switzerland) at 510 nm.
RT-PCR analysis
The preadipocyte 3 T3-L1 cells were plated onto 6-well plates on day 0 and incubated until confluency was achieved. For adipocyte differentiation, the confluent cells were treated with 1 μM dexamethasone, 1 μg/mL insulin, and 0.5 mM IBMX for 2 days, and the medium was replaced by fresh normal medium containing only 1 μg/mL insulin for 2 days. On day 2, the differentiated adipocyte cells were cultured in the presence or absence of EPK (25 or 50 μg/mL) for 6 days. The total RNA was extracted by using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA and subjected to PCR reaction by using a SuperScript One-Step reverse transcription-PCR (RT-PCR) kit (Invitrogen, Carlsbad, CA, USA). The PCR conditions were as follows: 30 cycles of 94 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s. The primer sequences have been provided in Table 1 (Additional file 2: Table S1). PCR products were run on 2 % agarose gels and then stained with ethidium bromide. Stained bands were visualized under UV light and photographed.
Western blot analysis
On day 2, the differentiated adipocyte cells were cultured in the presence or absence of EPK (25 or 50 μg/mL) or LA (200 μM) for 6 days. Cell were lysed in RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 % NP-40, 0.25 % deoxycholic acid-Na, 1 M EDTA, 1 mM Na3VO4, 1 mM NaF, and protease-inhibitor cocktail). The proteins in the samples were quantified using Bio-Rad DC protein assay kit II (Bio-Rad, Hercules, CA), separated by electrophoresis on 8 to 10 % SDS-PAGE gels, and electrotransferred onto a Hybond ECL transfer membrane (Amersham Pharmacia, Piscataway, NJ). The membranes were blocked in 3 % nonfat skim milk and probed with primary antibodies for PPARγ (Novus, Littleton, CO, USA), C/EBPα (Cell Signaling Tech., Danvers, MA, USA), p-AMPK (Cell Signaling Tech., MA, USA), AMPK (Cell Signaling Tech., MA, USA), adiponectin (Cell Signaling Tech., MA, USA), FABP (Cell Signaling Tech., Danvers, MA, USA), HMGCR (Bioss Antibodies, MA, USA), SREBP-1 (Sigma, St. Louis, MO, USA), or β-actin (Sigma, St. Louis, MO, USA) overnight. Subsequently, they were exposed to horseradish peroxidase (HRP)-conjugated secondary anti-mouse or rabbit antibodies. Protein expression was examined by using the enhanced chemiluminescence (ECL) system (Amersham Pharmacia, Piscataway, NJ).
AMPK gene silencing
AMPK small interfering RNA (siRNA) was purchased from Cell Signaling. A control siRNA was purchased from Santa Cruz Biotechnology. To transfect the siRNA, 3 T3-L1 cells were plated at a density of 4 × 105 cells per well in a 6-well plate. The cells were transfected using 100 nM of AMPK siRNA with INTERFERin (Poly plus, France) for 48 h. After treatment, the cells were confirmed by western blot.
HPLC analysis
In order to analyze the compounds from EPK, standards for the compounds were run on HICHROM HPLC columns (5 μM, 250 × 4.6 mm, Hichrom Ltd.) using a high HPLC system (Agilent Technologies, CA). The binary mobile phase consisted of 35 % to 100 % methanol-tetrahydrofuran solvent (99.5:0.5, v/v). The solvent flow rate was 1.0 mL/min and ambient temperature was set at 30 °C. UV detection was at a wavelength of 260 nm.
Animals
Male Sprague–Dawley rats (age, 4 weeks) were purchased from Hyo-Chang Science (Daegu, Korea). The rats were maintained under specific pathogen-free conditions with a 12 h light–dark cycle, 55 % humidity, and 22 ± 2 °C. All animal procedures were approved by the institutional Animal Care and Use Committee (IACUC) of Kyungsung University (Permit Number: 2011-13A), and performed in accordance with the Policy of the Ethical Committee of Ministry of Health and Welfare, Korea.
Experimental design and EPK treatment
Forty rats were divided into four groups (10 rats per group): normal group (low fat diet), control group (HFD), and two EPK-treated groups consuming HFD. Rats were fed the low fat diet or the HFD for 6 weeks. HFD composition has been described in Table 2. For EPK treatment dissolved in 4 % tween 80/normal saline was orally administered once daily to the rats at doses of 100 and 200 mg/kg for 6 weeks from the first day of HFD-feeding, whereas PBS was orally administered to the rats in the control group.
Preparation of rat serum
Whole blood was collected from rats by the cardiac puncture method and serum was isolated by centrifugation at 3000 rpm for 10 min.
Measurement of serum lipids level
Total cholesterol level was measured by using a total cholesterol assay kit (AM 202-K, Asan Pharm Co., Seoul, Korea) based on Richmond’s method [27]. Pipet 0.3 mL of the enzymatic cholesterol reagent into a test tube. Add either 20 μL of serum or glycerol standard. Mix well and incubate at 37 °C for at 30 min. Measure the absorbancesof samples and standard at 550 nm.
Triglyceride level was measured by a triglyceride assay kit (AM 157S-K, Asan Pharm Co., Seoul, Korea) based on McGowan’s method [28]. Pipet 0.3 mL of the enzymatic triglyceride reagent into a test tube. Add either 20 μL of serum or glycerol standard. Mix well and incubate at 37 °C for at 30 min. Measure the absorbancesof samples and standard at 500 nm.
Measurement of serum HDL and LDL levels
The levels of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) in serum were measured using the Roche Cobas C-111 analyzer (Roche-Diagnostics, Indianapolis, IN, USA): Atherosclerosis index (AI) was calculated by employing the following equation: AI = (total cholesterol − HDL cholesterol)/HDL cholesterol.
Measurement of body weight, retroperitoneal fat, and epididymal fat
The body weight of rats in normal (N), control (C), and EPK (100 and 200 mg/kg)-treated groups was monitored once a week for 6 weeks. The retroperitoneal and epididymal fat was also removed from EPK (100 and 200 mg/kg)-treated rats on the last day of animal study and weighed.
Immunohistochemical staining
For histopathological examination, paraffin sections (4 μM) from dissected liver and adipose tissues were stained with hematoxylin and eosin. Immunohistochemical staining of PPARγ (Novus, Littleton, CO, USA) and p-AMPK (Cell signaling Tech., MA, USA) was performed using the indirect avidin/biotin-enhanced horseradish peroxidase method. Antigen retrieval was performed after dewaxing and dehydration of the tissue sections by microwaving for 10 min in 10 mM citrate buffer. Sections were cooled to room temperature, treated with 3 % hydrogen peroxide in methanol for 10 min, and blocked with 6 % horse serum for 30 min at room temperature in a humidity chamber. The sections were then incubated with primary antibody against PPARγ (diluted 1: 200; Novus, Littleton, CO, USA) or p-AMPK (diluted 1: 150; Cell Signaling) at 4 °C overnight in a humidity chamber. The sections were washed in PBS and incubated with secondary antibody (biotinylated goat anti-rabbit antibody; diluted 1: 150; Vector Laboratories, Burlingame, CA, USA) for 30 min in the humidity chamber. After further washes, the antibodies were detected with the Vector ABC complex/horseradish peroxidase (HRP) kit (Vector Laboratories, Burlingame, CA, USA) and color developed with 3,3′-diaminobenzidine tetrahydrochloride. For semiquantitation, ten photomicrographs (200×) were taken with a CCD camera, avoiding gross necrotic areas.
Measurement of adipocyte size
Images were acquired using an Axio Imager. Adipocyte size in adipoxe tissue was analyzed using Image J (National Institutes of Health, Bethesda, MD) software.
Statistical analysis
All data are shown as mean ± SD. In vitro experiment data were analyzed by Student’s t-test. In vivo experiment data were calculated by analysis of variance (ANOVA) followed by Duncan’s multiple range test. A P value of less than 0.05 was considered statistically significant. Means in the same column with different superscript letters (a, b, c, d, e and f) are significantly different (P < 0.05) between groups.
Results
LA as an active ingredient in EPK
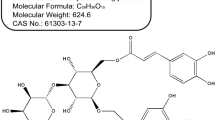
Through activity-guided fractionation (Additional file 1: Figure S1), we identified LA (Fig. 1a) from P. koraiensis needles as a novel anti-hyperlipidemic compound for use in increasing LDLR. The effects of LA on adipocyte differentiation and adipogenesis were assessed by western blotting and Oil-red-O staining. As shown in Fig. 1d, LA significantly decreased the expression of PPARγ, C/EBPα, related target genes (adiponectin, FAS, SREBP-1 and HMGCR) and increased the expression of p-AMPK, while no change was observed in its total protein expression. Furthermore, LA reduced lipid accumulation in 3 T3-L1 adipocytes by 32 % compared with that seen for the control group (Fig. 1b and c). To analyze LA in EPK, LA content was determined by HPLC. As shown in Fig. 1e, EPK contained LA. The peak showed that the retention time of LA was 59 min. The EPK included 98.7 μg/mg of LA.

Effects of LA on adipogenesis in 3 T3-L1 preadipocytes. a Chemical structure of LA. b 3 T3-L1 preadipocytes were treated with various concentrations of LA during differentiation. On day 8, the differentiated cells were stained with Oil-red-O dye and visualized under inverted microscopy at 100× magnification. c The cellular lipid retained Oil-Red-O in isopropanol and adipocyte expression was estimated by measuring O.D. with a microplate reader (Sunrise, TECAN, Mannedorf, Switzerland) at 510 nm. ### 0.001 < P (compared to preadipocte) and ** P < 0.01 (compared to adipocyte). Data are expressed as mean ± SD. d Total protein prepared from LA-treated 3 T3-L1 (preadipocytes or adipocyte) cells were subjected to western blot analysis for AMPK, p-AMPK, PPARγ, C/EBPα, adiponectin, FAS, SREBP-1, HMGCR, and β-actin. e HPLC analysis of LA in EPK. Preadipocyte (P)
Effect of EPK containing LA on fat accumulation in 3 T3-L1 adipocytes
Because in vitro study of adipose cells is easier in established cell lines [29], we examined the cytotoxic effect of EPK in 3 T3-L1 preadipocytes. Cells were treated with various concentrations of EPK (0, 3.125, 6.25, 12.5, 25, 50, 100, and 200 μg/mL) for 24 h, after which cell viability was evaluated by the MTT assay. As shown in Fig. 2a, EPK had no significant effect on the viability of 3 T3-L1 cells [23]. To investigate whether EPK alters the cellular differentiation in adipocytes, Oil-Red-O staining and triglyceride level analysis was performed in 3 T3-L1 cells [30]. 3 T3-L1 adipocytes were exposed to EPK for 6 days. The differentiated 3 T3-L1 adipocytes showed significantly increased fat accumulation (Fig. 2b and c) as compared to the control cells. EPK at concentrations of 25 or 50 μg/mL reduced the cellular lipid droplets in 3 T3-L1 adipocytes by 49 and 68.8 %, respectively, compared to the untreated control adipocytes. Moreover, EPK treatment reduced the level of serum triglycerides in 3 T3-L1 cells in a dose-dependent manner (21.8 and 33.7 % at concentrations of 25 and 50 μg/mL, respectively), compared to that in preadipocytes (59.92 %; Fig. 2d).

Effect of EPK on the differentiation of 3 T3-L1 adipocytes. a 3 T3-L1 cells were treated with various concentrations of EPK (0, 3.125, 6.25, 12.5, 25, 50, 100, and 200 μg/mL) for 24 h; then, the cell viability was evaluated by the MTT assay. b 3 T3-L1 preadipocytes were treated with various concentrations of EPK during differentiation. On day 8, the differentiated cells were stained with Oil-red-O dye and visualized under inverted microscopy at 100× magnification. c The cellular lipid retained Oil-Red-O in isopropanol and adipocyte expression was estimated by measuring O.D. with a microplate reader (Sunrise, TECAN, Mannedorf, Switzerland) at 510 nm. ### P < 0.001 (compared to preadipocyte) and *** P < 0.001 (compared to adipocyte). d Level of serumtriglyceride. ## P < 0.01 (compared to preadipocyte) and * P < 0.05 (compared to adipocyte). Data are expressed as mean ± SD. Preadipocyte (P)
Effect of EPK on adipogenesis in 3 T3-L1 preadipocytes
Adipocyte differentiation is the process of regulating a set of gene-expression events [31]. The well-studied PPARγ, C/EBPα, glycerol-3-phospate dehydrogenase (GPDH) and fatty acid-binding protein (FABP) are crucial transcription factor and promote adipogenesis [32]. To elucidate the regulatory mechanisms responsible for the anti-adipogenesis effect of EPK, the mRNA expression levels of PPARγ, C/EBPα, and GPDH were investigated by RT-PCR. EPK remarkably suppressed PPARγ, C/EBPα, and GPDH in 3 T3-L1 cells (Fig. 3a). In addition, the protein expression of adipogenic factors was examined using western blotting. As shown in Fig. 3b, the protein levels of PPARγ, C/EBPα, adiponectin SREBP-1, HMGCR, and FABP reduced in a concentration-dependent manner. AMPK is an important regulator of preadipocyte differentiation and adipogenesis. EPK at concentrations of 25 or 50 μg/mL increased p-AMPK, while no change was observed in its total protein expression (Fig. 3b). To further investigate the role of AMPK in preadipocyte differentiation and cholesterol synthesis and to confirm whether EPK modulates adipogenesis and cholesterol through AMPK, we used AMPK siRNA or the AMPK inhibitor compound C. We detected the expression of C/EBPα, PPARγ, FAS, SREBP-1, and HMGCR after cells were treated with the AMPK inhibitor compound C or AMPK siRNA. As shown in Fig. 3c and d, AMPK siRNA significantly reduced AMPK protein expression. The p-AMPK induced by EPK was abrogated by pretreatment with compound C or AMPK siRNA. PPARγ, FAS, SREPBP-1, and HMGCR, downstream of AMPK, were decreased by EPK treatment and these effects were reversed in the presence of compound C or AMPK siRNA. In addition, the lipid content of fat droplets was also restored in the presence of compound C (Fig. 3e).

Effect of EPK on adipogenesis in 3 T3-L1 preadipocytes. 3 T3-L1 preadipocytes were incubated in medium containing insulin (1.0 μg/mL) with or without the indicated concentrations of EPK or CC. a Total RNA was extracted from 3 T3-L1cells treated with EPK and used for RT-PCR analysis of PPARγ, C/EBPα, GPDH, and β-actin. b Total proteins prepared from EPK-treated 3 T3-L1 cells were subjected to western blot analysis for AMPK, p-AMPK, PPARγ, C/EBPα, adiponectin, FABP, SREBP-1, HMGCR, and β-actin. c 3 T3-L1 cells were transfected with AMPK-siRNA for 48 h and treated with or without EPK (50 μg/mL) for 24 h and subjected to western blot analysis for AMPK, p-AMPK, PPARγ, SREBP-1, and β-actin. d Total proteins prepared from EPK (50 μg/mL) or CC (2 μM)-treated 3 T3-L1 cells for 6 h were subjected to western blot analysis for AMPK, p-AMPK, PPARγ, FAS, SREBP-1, HMGCR, and β-actin. e Cells were fixed and stained with Oil-Red-O. The Oil-Red-O-stained adipocytes were photographed at 100× magnification under a microscope. Preadipocyte (P), compound C (CC)
Effect of EPK on the body weight of HFD-fed rats
The body weights of normal group (N), control group (C), and EPK-treated groups were monitored once a week for 6 weeks. As shown in Fig. 4a, the body weights of the HFD-fed control group were significantly greater at 2 weeks after feeding, compared to those for the normal low fat group. In contrast, the body weight gain was dose dependently abrogated in EPK-treated groups, from the 3rd week of treatment with EPK. In addition, 6 weeks after treatment, body weights increased to 331.4 ± 2.8 g in the HFD-fed control group (normal group, 281.6 ± 1.7 g). However, EPK significantly suppressed the body weights to 309.5 ± 1.8 g and 303.7 ± 2.0 g, respectively, at doses of 100 mg/kg and 200 mg/kg.

Effect of EPK on body and abdominal fat weights of HFD-fed rats. Rats fed a HFD were orally treated with or without EPK daily for 6 consecutive weeks. a Body weights of rats. b Abdominal fat and retroperitoneal and epididymal fat from rats treated with or without EPK (100 and 200 mg/kg) were removed and weighed. c Average size of adipocytes measured in 5 different fields. ###P < 0.001 (compared to the normal), **P < 0.01 and ***P < 0.001 (compared to the control group). d Representative picture of immunohistochemical staining for PPARγ and p-AMPK in adipocyte tissue sections. ###P < 0.001 (compared to normal group), **P < 0.01 and ***P < 0.001 (compared to control group). e Representative picture of immunohistochemical staining for PPARγ and p-AMPK in liver tissue sections. ##P < 0.01 (compared to normal group), **P < 0.01 and *** P < 0.001 (compared to control group). Data are expressed as mean ± SD. Values with the different superscript letters indicate statistical significance (P < 0.05) between groups by Duncan’s multiple range test. Normal group (N), control group (C)
Effects of EPK on abdominal fat weight of HFD-fed rats
The retroperitoneal and epididymal fat weights were measured to test whether the body weight normalization by EPK in HFD-fed rats was associated with reduction of fat content in the body. In this study, HFD significantly increased the retroperitoneal fat weight to 18.3 ± 2.91 mg/g from 6.21 ± 1.26 mg/kg body weight. In contrast, EPK decreased the retroperitoneal fat weight to 15.7 ± 1.86 and 12.7 ± 1.43 mg/g at doses of 100 and 200 mg/kg, respectively, compared to the HFD-fed control group (Fig. 4b). Likewise, epididymal fat weight also increased in the HFD-fed rats compared to the weight in the normal group. Oral administration of EPK decreased epididymal fat pad weight by 10.9 ± 1.85 and 8.52 ± 1.96 mg/g at doses of 100 and 200 mg/kg, respectively (Fig. 4b). Moreover, HFD-fed rats showed a significant increase (53.1 %, P < 0.01) in the cell size of epididymal adipose tissues compared to normal adipose tissues, whereas EPK-treated groups showed significantly decreased hypertrophic adipocyte formation (Fig. 4c). The expression of p-AMPK in both adipose and liver tissues was increased by EPK in the HFD compared to the control group. In addition, the expression of PPARγ in both adipose and liver tissue was lower in the EPK treated group than in the control group (Fig. 4d and e).
Effects of EPK on serum lipid and cholesterol levels in HFD-fed rats
The consumption of the HFD significantly increased triglyceride levels in the control group, as compared to those in the normal low fat group (Fig. 5a). EPK treatment decreased the level of serum triglyceride from 131.8 ± 18.2 to 109.3 ± 11.5 and 94.2 ± 8.43 mg/dL at 100 and 200 mg/kg, respectively (Fig. 5a). Additionally, the consumption of HFD significantly increased serum total cholesterol compared to that in the normal low fat group, while EPK significantly reduced the level of total cholesterol in a dose-dependent manner (Fig. 5b). The intake of the HFD significantly decreased the level of HDL but increased the level of LDL compared to those for the normal group (Fig. 5c, control). However, EPK did not have a significant impact on LDL levels but elevated HDL levels in a dose-dependent manner compared to those seen for the HFD control group (Fig. 5c). EPK treatment significantly decreased the AI value in a dose-dependent manner compared to the AI determined for the HFD control (Fig. 5d).

Effects of EPK on serum lipid and cholesterol levels and on hepatic triglyceride and cholesterol. a Triglyceride levels were measured with a triglyceride assay kit (AM157S-K, Asan PharmCo., Seoul, Korea). b Total cholesterol levels were measured by using a total cholesterol assay kit (AM202-K, Asan PharmCo., Seoul, Korea). c The levels of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) in serum were measured using the Cobas c 111 analyzer (Roche-Diagnostics, Indianapolis, IN, USA). d AI was calculated by employing the following equation: AI = (total cholesterol − HDL cholesterol)/HDL cholesterol. e The levels of triglyceride in liver were measured with the Biochemistry Analyzer. f The levels of total cholesterol in the liver were measured with the Biochemistry Analyzer. Data are expressed as mean ± SD. Values with the different superscript letters indicate statistical significance (P < 0.05) between groups by Duncan’s multiple range test. Normal group (N), control group (C)
Effects of EPK on hepatic triglyceride and cholesterol levels in HFD-fed rats
The consumption of HFD significantly increased hepatic triglyceride (Fig. 5e) and hepatic cholesterol levels (Fig. 5f), as compared to those in the normal low fat group. Oral treatment with EPK reduced triglyceride levels in the liver in a dose-dependent manner (24.4 ± 2.47 and 21.2 ± 1.88 mg/g at doses of 100 and 200 mg/kg, respectively), compared to those for the control group (29.8 ± 3.25 mg/g tissue) (Fig. 5e). In addition, EPK administration significantly lowered hepatic cholesterol levels in the liver from 16.9 ± 2.18 mg/g in the HFD control group to 12.6 ± 1.42 and 10.4 ± 1.59 mg/g at 100 and 200 mg/kg, respectively (Fig. 5f).
Discussion
This study provides the first direct evidence of the anti-obesity effects of EPK and its component LA, while providing insight into the regulatory mechanisms underlying those effects in rats with HFD-induced obesity. Our findings show that EPK efficaciously inhibits adipocyte differentiation and adipogenesis in 3 T3-L1 adipocytes and in rats with HFD-induced obesity by activating AMPK. In recent years, EOPK has been reported to demonstrate anti-cancer, anti-obesity, and anti-hypolipidemic effects [21, 23, 24]. EPK is easier to extract than EOPK. There are significant differences between EPK and EOPK with respect to their components and extraction cost of the two extracts. Moreover, EPK is convenient and easy to use.
Adipogenesis is a cellular differentiation process in which the preadipocytes are transformed into differentiated adipocytes [31] and accumulate lipids [33]. Therefore, controlling adipocyte differentiation is important. AMPK agonists and PPARγ antagonists appear to be involved in adipocyte differentiation and thus can be potential drugs for the treatment of obesity [34]. EPK suppressed fat accumulation and serum triglyceride levels, decreased PPARγ, CEBPα, FABP, and GPDH expression, and increased p-AMPK expression in the differentiated 3 T3-L1 adipocytes, without any cytotoxic effect. Similarly, many natural products, including green tomato extract [35], tiacremonone [4, 36, 37], and ursolic acid [38], suppress adipogenesis and improve insulin sensitivity in vitro and in vivo via AMPK and PPARγ signaling. Moreover, EPK in HFD-fed SD rats was found to reduce body weight gain without loss of appetite (Additional file 3: Figure S2). Loss of body weight is related to decrease in fat pad mass as a result of reduction of adipocyte size or triglyceride accumulation [39]. EPK reduces the retroperitoneal and epididymal fat weight as well as serum triglyceride levels compared to those in HFD-fed rats. Our data suggest that EPK can prevent obesity via inhibition of lipid metabolism, including reduction of triglyceride levels.
Elevated cholesterol is a risk factor for obesity, stroke, and heart disease, as well as diabetes [40]. Specifically, low levels of serum HDL cholesterol are closely related to obesity [41]. EPK increased the level of HDL cholesterol in a dose-dependent manner compared to that in HFD-fed rats. Furthermore, EPK decreased triglyceride and cholesterol levels, suggesting that EPK regulates lipid metabolism, which was in accordance with in vitro data. EPK inhibited SREBP-1 and HMGCR as the cholesterol related factor. Adipogenesis and cholesterol syntheses in adipocytes as well as the liver, is regulated by SREBP-1 and HMGCR [20, 42, 43]. Several natural compounds including mevalonic acid [42], suppressed cholesterol levels in vitro and in vivo through AMPK, SREBP-1, and HMGCR in adipocytes, similary to our data. LA as an active compound in EPK that is responsible for anti-hyperlipidemic activity was identified by a bioactivity-guided fractionation procedure. The active fractions were characterized by reverse-phase HPLC and identified by mass spectrometry (Additional file 1: Figure S1). LA is a bioactive diterpene and is known to have anti-allergic effects [44]. LA inhibited fat accumulation in adipocytes, induced p-AMPK expression, and inhibited PPARγ expression. Further study is required to verify the anti-obesity effects of LA in HFD-fed animal models.
Conclusion
In summary, EPK decreased fat accumulation, serum triglyceride levels, and PPARγ, CEBPα, FABP, GPDH, SREBP-1, and HMGCR expression and induced p-AMPK in the differentiated 3 T3-L1 adipocytes (Fig. 6). Furthermore, EPK reduced the serum and hepatic levels of triglyceride and total cholesterol in an in vivo model. LA as an active compound in EPK inhibited adipogenesis and controlled cholesterol synthesis related proteins level in 3 T3-L1 adipocytes via AMPK pathways. Overall, our findings suggest the potential of EPK as an anti-obesity and anti-hyperlipidemia agent.

Effects of EPK and LA on anti-obesity in 3 T3-L1-
Abbreviations
- EPK:
-
Ethanol extract of Pinus koraiensis
- PPARγ:
-
Peroxisome proliferator-activated receptor-γ
- C/EBPα:
-
CCAAT/enhancer-binding proteins α
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- LA:
-
Lambertianic acid
References
Steinberger J, Daniels SR, American Heart Association Atherosclerosis H, Obesity in the Young C, American Heart Association Diabetes C. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: an American Heart Association scientific statement from the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism). Circulation. 2003;107(10):1448–53.
Wing RR, Jeffery RW. Effect of modest weight loss on changes in cardiovascular risk factors: are there differences between men and women or between weight loss and maintenance? Int J Obes Relat Metab Disord. 1995;19(1):67–73.
Caro JF, Dohm LG, Pories WJ, Sinha MK. Cellular alterations in liver, skeletal muscle, and adipose tissue responsible for insulin resistance in obesity and type II diabetes. Diabetes Metab Rev. 1989;5(8):665–89.
Huang B, Yuan HD, Kim do Y, Quan HY, Chung SH. Cinnamaldehyde prevents adipocyte differentiation and adipogenesis via regulation of peroxisome proliferator-activated receptor-gamma (PPARgamma) and AMP-activated protein kinase (AMPK) pathways. J Agric Food Chem. 2011;59(8):3666–73.
Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14(11):1293–307.
Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–56.
Zuo Y, Qiang L, Farmer SR. Activation of CCAAT/enhancer-binding protein (C/EBP) alpha expression by C/EBP beta during adipogenesis requires a peroxisome proliferator-activated receptor-gamma-associated repression of HDAC1 at the C/ebp alpha gene promoter. J Biol Chem. 2006;281(12):7960–7.
Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996;10(9):1096–107.
Penque BA, Hoggatt AM, Herring BP, Elmendorf JS. Hexosamine biosynthesis impairs insulin action via a cholesterolgenic response. Mol Endocrinol. 2013;27(3):536–47.
Fajas L, Schoonjans K, Gelman L, Kim JB, Najib J, Martin G, et al. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol. 1999;19(8):5495–503.
Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546(1):113–20.
Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25.
Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217–22.
Hardie DG, Corton J, Ching YP, Davies SP, Hawley S. Regulation of lipid metabolism by the AMP-activated protein kinase. Biochem Soc Trans. 1997;25(4):1229–31.
Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30(Pt 6):1064–70.
Chen S, Li Z, Li W, Shan Z, Zhu W. Resveratrol inhibits cell differentiation in 3T3-L1 adipocytes via activation of AMPK. Can J Physiol Pharmacol. 2011;89(11):793–9.
Lee YK, Lee WS, Hwang JT, Kwon DY, Surh YJ, Park OJ. Curcumin exerts antidifferentiation effect through AMPKalpha-PPAR-gamma in 3T3-L1 adipocytes and antiproliferatory effect through AMPKalpha-COX-2 in cancer cells. J Agric Food Chem. 2009;57(1):305–10.
Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276(41):37731–4.
Le Lay S, Lefrere I, Trautwein C, Dugail I, Krief S. Insulin and sterol-regulatory element-binding protein-1c (SREBP-1C) regulation of gene expression in 3T3-L1 adipocytes. Identification of CCAAT/enhancer-binding protein beta as an SREBP-1C target. J Biol Chem. 2002;277(38):35625–34.
Chiu CY, Chan IL, Yang TH, Liu SH, Chiang MT. Supplementation of chitosan alleviates high-fat diet-enhanced lipogenesis in rats via adenosine monophosphate (AMP)-activated protein kinase activation and inhibition of lipogenesis-associated genes. J Agric Food Chem. 2015;63(11):2979–88.
Kim JH, Lee HJ, Jeong SJ, Lee MH, Kim SH. Essential oil of Pinus koraiensis leaves exerts antihyperlipidemic effects via up-regulation of low-density lipoprotein receptor and inhibition of acyl-coenzyme A: cholesterol acyltransferase. Phytother Res. 2012;26(9):1314–9.
Joo HE, Lee HJ, Sohn EJ, Lee MH, Ko HS, Jeong SJ, et al. Anti-diabetic potential of the essential oil of Pinus koraiensis leaves toward streptozotocin-treated mice and HIT-T15 pancreatic beta cells. Biosci Biotechnol Biochem. 2013;77(10):1997–2001.
Ko HS, Lee HJ, Sohn EJ, Yun M, Lee MH, Kim SH. Essential Oil of Pinus koraiensis Exerts Antiobesic and Hypolipidemic Activity via Inhibition of Peroxisome Proliferator-Activated Receptors Gamma Signaling. Evid Based Complement Alternat Med. 2013;2013(2013):947037.
Cho SM, Lee EO, Kim SH, Lee HJ. Essential oil of Pinus koraiensis inhibits cell proliferation and migration via inhibition of p21-activated kinase 1 pathway in HCT116 colorectal cancer cells. BMC Complement Altern Med. 2014;14:275.
Asset G, Staels B, Wolff RL, Bauge E, Madj Z, Fruchart JC, et al. Effects of Pinus pinaster and Pinus koraiensis seed oil supplementation on lipoprotein metabolism in the rat. Lipids. 1999;34(1):39–44.
Ferramosca A, Savy V, Conte L, Zara V. Dietary combination of conjugated linoleic acid (CLA) and pine nut oil prevents CLA-induced fatty liver in mice. J Agric Food Chem. 2008;56(17):8148–58.
Richmond W. Use of cholesterol oxidase for assay of total and free cholesterol in serum by continuous-flow analysis. Clin Chem. 1976;22(10):1579–88.
McGowan MW, Artiss JD, Strandbergh DR, Zak B. A peroxidase-coupled method for the colorimetric determination of serum triglycerides. Clin Chem. 1983;29(3):538–42.
Poulos SP, Dodson MV, Hausman GJ. Cell line models for differentiation: preadipocytes and adipocytes. Exp Biol Med. 2010;235(10):1185–93.
Yang J, Ren J, Song J, Liu F, Wu C, Wang X, et al. Glucagon-like peptide 1 regulates adipogenesis in 3T3-L1 preadipocytes. Int J Mol Med. 2013;31(6):1429–35.
Otto TC, Lane MD. Adipose development: from stem cell to adipocyte. Crit Rev Biochem Mol Biol. 2005;40(4):229–42.
Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7(12):885–96.
Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12(11):722–34.
Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008;582(1):81–9.
Choi KM, Lee YS, Shin DM, Lee S, Yoo KS, Lee MK, et al. Green tomato extract attenuates high-fat-diet-induced obesity through activation of the AMPK pathway in C57BL/6 mice. J Nutri Biochem. 2013;24(1):335–42.
Ban JO, Lee DH, Kim EJ, Kang JW, Kim MS, Cho MC, et al. Antiobesity effects of a sulfur compound thiacremonone mediated via down-regulation of serum triglyceride and glucose levels and lipid accumulation in the liver of db/db mice. Phytother Res. 2012;26(9):1265–71.
Kim EJ, Lee DH, Kim HJ, Lee SJ, Ban JO, Cho MC, et al. Thiacremonone, a sulfur compound isolated from garlic, attenuates lipid accumulation partially mediated via AMPK activation in 3T3-L1 adipocytes. J Nutr Biochem. 2011;23(12):1552–8.
He Y, Li Y, Zhao T, Wang Y, Sun C. Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through LKB1/AMPK pathway. PLoS One. 2013;8(7), e70135.
Rosenbaum M, Leibel RL, Hirsch J. Obesity. N Eng J Med. 1997;337(6):396–407.
Rashid S, Genest J. Effect of obesity on high-density lipoprotein metabolism. Obesity. 2007;15(12):2875–88.
Woods SC, Seeley RJ, Porte Jr D, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280(5368):1378–83.
Liu X, You W, Cheng H, Zhang Q, Song E, Wan F, et al. Effect of mevalonic acid on cholesterol synthesis in bovine intramuscular and subcutaneous adipocytes. J Appl Genet. 2016;57(1):113–8.
Kim SJ, Jung JY, Kim HW, Park T. Anti-obesity effects of Juniperus chinensis extract are associated with increased AMP-activated protein kinase expression and phosphorylation in the visceral adipose tissue of rats. Biol Pharm Bull. 2008;31(7):1415–21.
Chae HS, Chin YW. Anti-allergic effect of lambertianic acid from Thuja orientalis in mouse bone marrow-derived mast cells. Immunopharmacol Immunotoxicol. 2012;34(2):250–5.
Acknowledgment
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2007–0054931).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that there are no conflicts of interest.
Authors’ contributions
HJL conceived and designed the experiments; SMC and MSL performed the experiments; EOL analyzed the data; and MHL and SHK contributed reagents/analysis tools. All authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
Flow diagram for the extraction and fractionation of LA from P. koraiensis. (TIF 2837 kb)
Additional file 2: Table S1.
Composition of fat in HFD. (PDF 7 kb)
Additional file 3: Figure S2.
Food intake. (TIF 2565 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, MS., Cho, SM., Lee, Mh. et al. Ethanol extract of Pinus koraiensis leaves containing lambertianic acid exerts anti-obesity and hypolipidemic effects by activating adenosine monophosphate-activated protein kinase (AMPK). BMC Complement Altern Med 16, 51 (2016). https://doi.org/10.1186/s12906-016-1031-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-016-1031-2