
Abstract
Background
Periodontitis is strongly associated with type 2 diabetes (T2D) that results in serious complications and mortality. However, the pathogenic role of periodontitis in the development of T2D and the underlain mechanism have not been fully elucidated.
Methods
A Mendelian randomization (MR) was performed to estimate the causality between two diseases. Bioinformatics tools, including gene ontology and pathway enrichment analyses, were employed to analyze the common differentially expressed genes (DEGs) in periodontitis and T2D. MR and colocalization analyses were then utilized to investigate the causal associations between potential pathogenic gene expression and the risk of T2D. Single cell-type expression analysis was further performed to detect the cellular localization of these genes.
Results
Genetically predicted periodontitis was associated with a higher risk of T2D (OR, 1.469; 95% CI, 1.117–1.930; P = 0.006) and insulin resistance (OR 1.034; 95%CI 1.001–1.068; P = 0.041). 79 common DEGs associated with periodontitis and T2D were then identified and demonstrated enrichment mainly in CXC receptor chemokine receptor binding and interleutin-17 signaling pathway. The integration of GWAS with the expression quantitative trait locis of these genes from the peripheral blood genetically prioritized 6 candidate genes, including 2 risk genes (RAP2A, MCUR1) and 4 protective genes (WNK1, NFIX, FOS, PANX1) in periodontitis-related T2D. Enriched in natural killer cells, RAP2A (OR 4.909; 95% CI 1.849–13.039; P = 0.001) demonstrated high risk influence on T2D, and exhibited strong genetic evidence of colocalization (coloc.abf-PPH4 = 0.632).
Conclusions
This study used a multi-omics integration method to explore causality between periodontitis and T2D, and revealed molecular mechanisms using bioinformatics tools. Periodontitis was associated with a higher risk of T2D. MCUR1, RAP2A, FOS, PANX1, NFIX and WNK1 may play important roles in the pathogenesis of periodontitis-related T2D, shedding light on the development of potential drug targets.
Similar content being viewed by others

Background
Periodontitis, a significant contributor to the overall global disease burden, represents a public health problem [1]. Increased by 8.44% (6.62-10.59%) in just 20 years, the age-standardized prevalence rate of periodontitis reached 13,109 (9,993-16,385) per 100,000 people in 2019 [2]. Epidemiologic studies indicating a correlation between periodontitis and at least 43 systemic diseases, including type 2 diabetes (T2D) [3,4,5,6,7]. T2D is associated with serious complications and mortality, resulting in low quality of life and substantial socioeconomic burden [8]. Periodontitis patients have been reported to have a higher risk of T2D, with severe periodontitis increasing the risk of T2D by 53% [9,10,11]. Another study indicates that moderate or severe periodontitis is related to a higher risk of all-cause mortality in diabetes patients [12].
Previous studies indicated that periodontitis may contribute to T2D development due to systemic microinflammation [11, 13]. The microbiome associated with periodontitis may activate the host immune response, leading to the secretion and cascade amplification of numerous pro-inflammatory cytokines, thereby promoting the progression of T2D [14]. Moreover, immune cells exhibited similar functional alterations in T2D and periodontitis [15]. A notable pathway RESISTIN, which could increase insulin resistance and susceptibility to diabetes, was found to be activated under periodontitis conditions, potentially linking periodontitis to T2D [16]. Despite the findings of these studies, there remains limited research confirming causally that periodontitis can precipitate the onset of diabetes. Moreover, the specific molecular mechanisms through which the key pathogenic genes mediate periodontitis-related T2D remain largely ambiguous.
Mendelian randomization (MR) emerges as an alternative method to randomized clinical trial, allowing for the exploration of potential causality between an exposure and the outcome by employing use of genetic variants as instrumental variables (IVs) [17]. Similar to randomization in randomized clinical trial, genetic variants are randomly assigned during gamete formation and conception, thus minimizing the impact of confounders and reverse causation. The expression quantitative trait loci (eQTL) can reveal the associations of single nucleotide polymorphisms (SNPs) with level of gene expression. The increasing availability of eQTL data enables the exploration of causal association between gene expression and traits via MR [18], allowing the identification of key pathogenic genes in diseases.
In this study, a two-sample MR analysis is conducted to illustrate the relationship between periodontitis and T2D. Multiple bioinformatics tools are utilized to identify the differentially expressed genes (DEGs) and elucidate the potential mechanism underlying periodontitis-related T2D. Subsequently, MR is employed to explore the associations between pathogenic gene expression and the risk of T2D. Colocalization analysis is employed to provide additional evidence supporting causality. Lastly, single-cell expression analysis is conducted to investigate the cellular localization of these genes. Through these methodologies, this study aims to deepen the understanding of the role of periodontitis in the development of T2D, offering new perspectives for T2D management.
Methods
Overall study design
The framework of the study is depicted in Fig. 1. Briefly, two-sample MR was employed to explore the causal association between periodontitis and T2D. To discover the mechanisms of periodontitis-related T2D, common DEGs related to periodontitis and T2D were identified and analyzed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis. Then, eQTL data were employed to examined the associations between DEGs expression levels and T2D using a MR framework. Colocalization analysis was leveraged to further verify the causal associations between the candidate genes and T2D. Last, single cell-type expression analysis was conducted to investigate the cellular localization of candidate genes in peripheral blood mononuclear cells (PBMCs) of patients with T2D and periodontitis.

The framework of the study. DEGs indicates differentially expressed genes; T2D indicates type 2 diabetes
GWAS data sources of periodontitis, T2D and eQTL
For the analysis of periodontitis, the genome-wide association study (GWAS) summary statistics of periodontitis in European ancestry that includes 4,434 periodontitis cases and 259,234 controls from FinnGen were selected (Supplementary Table 1). Details of this project could be found in the FinnGen research project (https://www.finngen.fi/fi). The International Classification of Diseases, Tenth Revision (ICD-10) was used to define the periodontitis cases. The severity of periodontitis was not reported in the database. Regarding T2D, the GWAS summary statistics of Europeans were obtained from the DIAGRAM Consortium (http://www.diagram-consortium.org/) and the IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/). European ancestry specific dataset from the DIAGRAM Consortium (180,834 T2D cases and 1,159,055 controls) was included in the discovery MR analysis [19], and T2D dataset from IEU OpenGWAS (4,040 T2D cases and 116,246 controls) was further analyzed in the validation MR. The GWAS statistics of the level of fasting insulin (FI) were obtained from the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) (http://magicinvestigators.org/), which included 281,416 individuals without diabetes. All cited data sources obtained participant informed consent and relevant ethical approval. The cis-eQTL data from whole blood and PBMC tissues were extracted from eQTLGen Consortium (https://www.eqtlgen.org/) which included 14,263 individuals [20].
Selection of genetic variants as IVs
The SNPs associated with periodontitis were selected at the genome-wide significance of P < 5 × 10− 6 [21]. The independence of the IV was ensured via the linkage disequilibrium (LD) clumping algorithm with the stringent cut-off r2 = 0.001. Proxy-SNPs were not used as IVs when there were no matching SNPs in the outcome data. To ascertain the independence assumption, SNPs were checked in PhenoScanner V2 (http://www.phenoscanner.medschl.cam.ac.uk/) to test if these SNPs were correlated with the potential cofounding factors [22]. Any SNPs found to be associated with any of the confounders were subsequently excluded from this study. F statistic was used to determine the power of the remaining SNPs, and the SNP with an F statistic < 10 was removed. The F statistic was associated with the proportion of variance in the phenotype explained by the genetic variants (R2), sample size (n) and number of instruments (k) and calculated by the formula (1) and (2) [23].
Mendelian randomization
The TwoSampleMR and MendelianRandomization packages were mainly used to perform MR analyses [24, 25]. The inverse-variance-weight (IVW) method was the primary method employed for analysis. Heterogeneity in IVW analysis was estimated by using the Cochran Q statistic. Specifically, a random-effects IVW MR analysis should be used if Cochran Q statistic indicates heterogeneity [26]. The presence of pleiotropy was estimated using MR-Egger regression based on its intercept term [27, 28]. The slope coefficient from MR-Egger regression was utilized for evaluating causal effect when pleiotropy was detected. The pleiotropy was also assessed through MR pleiotropy residual sum and outlier (MR-PRESSO) that is designed to identify horizontal pleiotropic outliers [29]. Weighted median, Simple mode and Weight mode analyses are performed as sensitivity analyses to bolster the robustness of the findings of IVW method [30, 31]. The sensitivity of SNPs was tested through leave-one-out analysis that performing multiple analyses after sequentially removing 1 SNP from the IVs set. Moreover, the impact of each SNP on the outcome was visualized in forest plots. The funnel plots were also used to assess potential biases in the results. The MR study adheres to the recommendations by the Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization (STROBE-MR) reporting guideline [32].
Collection of periodontitis and T2D related genes
The microarray dataset consisted of 9 T2D whole-blood samples and 9 healthy whole-blood samples (GSE189005) and dataset consisted of 183 periodontitis site samples and 64 healthy periodontal site samples (GSE10334) were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) [33]. The limma package was used to screen the DEGs in datasets [34]. DEGs were identified upon the thresholds of adjusted P value < 0.05 and |log2(fold change)|>0.5. Subsequently, the expression patterns of DEGs were visualized in the form of volcano plot and heatmap with the ggplot2 and pheatmap packages. The common DEGs of periodontitis and T2D were then overlapped for further analyses.
Functional enrichment analysis
To understand the functional characteristic of the common DEGs in periodontitis and T2D, GO and KEGG pathway enrichment analysis were then conducted by ClusterProfiler package [35]. A threshold of P < 0.05 was defined as significant enrichment. The results of functional enrichment analysis were displayed via bar plot.
Colocalization analysis
To assess whether causal gene and T2D risk were consistent with a shared causal variant, the colocalization analyses were conducted based on coloc and locuscomparer R packages [36, 37]. For each gene, SNPs within ± 1000,000 kb of the eQTL were included. When a gene had more than one eQTL, colocalization analysis was performed based on the eQTL with the most significant P value. The eQTL with the strongest evidence for colocalization was shown in the regional association plots. PPH4 > 0.6 was defined as the threshold for the shared genetic effects between the two traits [38].
Single cell-type expression analysis
The cell type-specific expression of causal genes was further evaluated by employing single-cell data of PBMCs from patients with periodontitis and T2D (GSE244515) from GEO database [33]. Data preprocessing was carried out by Seurat and Harmony packages [39, 40]. The SingleR package was used to annotate the cell types [41], and the clustering results were visualized using uniform manifold approximation and projection (UMAP).
Results
Identification of causal association between periodontitis and T2D
To explore the causality between periodontitis and T2D, MR analysis was performed (Fig. 2A). As presented in Supplementary Table 2, 17 independent significant SNPs nominally related to periodontitis were selected. IVW method was conducted as the primary analysis, demonstrating that T2D was causally associated with an increased risk of periodontitis (OR, 1.045; 95% CI, 1.012–1.079; P = 0.007). However, the MR-Egger, Weighted median, Simple mode and Weight mode analyses did not present significant associations between periodontitis and T2D (Fig. 2B). Sensitivity analyses revealed no evidence of heterogeneity (Q statistic = 18.89; P = 0.219) or pleiotropy (MR-Egger intercept P = 0.772). The MR-PRESSO global outlier test did not identify a significant impact of outliers (P = 0.265). Another statistics was analyzed to test the robustness of the conclusion. For this dataset, the IVW leave-one-out analysis showed that rs141098993 was an outliner. After excluding rs141098993, genetically predicted periodontitis was causally associated with a higher risk of T2D (OR, 1.469; 95% CI, 1.117–1.930; P = 0.006) (Fig. 2C), further substantiating the causal relationship. Since insulin resistance was an initial abnormality in the development of T2D, relationship between periodontitis and FI, the biochemical parameters that reflected the β-cell insulin production and insulin resistance, was further analyzed to confirm the conclusion [42, 43]. The IVW analysis results indicated that periodontitis was associated with increased level of FI (OR 1.034; 95%CI 1.001–1.068; P = 0.041) (Fig. 2D). The results were corroborated by the weight median method (OR 1.041; 95%CI 1.003–1.080; P = 0.033), indicating reliability of the conclusion. The IVW leave-one-out analysis, scatter plot, forest plot, and funnel plot did not show any leverage points with high influence (Supplementary Figs. 1–3).

MR results indicate the relationship between periodontitis and T2D. (A) The basic principles of the MR study. (B) MR estimates for the impact of periodontitis on T2D (discovery phase). (C) MR estimates for the influence of periodontitis on T2D (validation phase). (D) MR estimates for the impact of periodontitis on FI. IV indicates instrumental variables; OR indicates odds ratio; CI indicates confidence interval. The error bars represent 95%CIs. All statistical tests were two-sided. P < 0.05 was considered significant
Functional enrichment of DEGs involved in periodontitis-related T2D
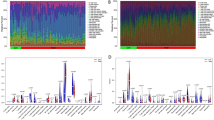
To explore the genes involved in the pathogenesis of periodontitis-related T2D, expression profiles of periodontitis and T2D from the GEO database were analyzed. In the periodontitis dataset, 1,462 DEGs were collected with |Log2(fold change)|>0.5 and |adj.p.Val.|<0.05, and 2,458 DEGs were identified in the T2D dataset (Fig. 3A-D). Venn diagram revealed that 79 DEGs were involved in both periodontitis and T2D (Fig. 3E). The GO and KEGG enrichment analyses were performed to better understand the function and mechanism of the pathogenic genes. Biological process (BP) of GO term analysis demonstrated that the pathogenic genes in periodontitis-related T2D were mostly enriched in response to lipopolysaccharide and molecule of bacterial origin. In terms of cellular component (CC) of GO term analysis, the pathogenic genes were mainly located in platelet alpha granule and secretory granule lumen. For molecular function (MF) analysis, the results suggested that CXC receptor chemokine receptor binding and chemokine activity were the most relevant items of the pathogenic genes. KEGG pathway analysis indicated that the pathogenic genes in periodontitis-related T2D were associated with interleutin-17 (IL-17) signaling pathway and malaria (Fig. 3E).

Identification of DEGs in periodontitis-related T2D, and followed by enrichment analyses. (A) The volcano plot revealing DEGs in the T2D dataset. (B) The heatmap representing the DEGs in the T2D dataset. (C) The volcano plot revealing DEGs in the periodontitis dataset. (D) The heatmap representing the DEGs in the periodontitis dataset. (E) The intersection of T2D DEGs with periodontitis DEGs via the Venn diagram. (F) The GO/KEGG enrichment analyses of pathogenic genes in periodontitis-related T2D. T2D indicates type 2 diabetes; PBMC indicates peripheral blood mononuclear cells; DEG indicates differentially expressed genes
Identification of T2D causal genes
To further investigate the molecular mechanisms, eQTLs of the pathogenic genes from the blood were obtained to perform MR from eQTLGen Consortium. As a result, 6 potential candidate genes were identified to have causal associations with T2D (Fig. 4A). Mitochondrial calcium uniporter regulator 1 (MCUR1) (OR 2.836; 95% CI 1.263–6.367; P = 0.016) and RAS-related protein Rap-2a (RAP2A) (OR 4.909; 95% CI 1.849–13.039; P = 0.001) were up-regulated in T2D-PBMCs and demonstrated risk influence on T2D. FBJ murine osteosarcoma viral oncogene homolog (FOS) (OR 0.479; 95% CI 0.248–0.926; P = 0.029) was down-regulated in T2D-PBMCs, demonstrating protective influence on T2D. However, nuclear factor IX (NFIX) (OR 0.420; 95% CI 0.207–0.850; P = 0.016), pannexin 1 (PANX1) (OR 0.839; 95% CI 0.744–0.947; P = 0.004) and with no lysine-1 (WNK1) (OR 0.265; 95% CI 0.102–0.689; P = 0.006) were up-regulated in T2D-PBMCs and showed protective influence on T2D (Fig. 4B-C).

MR analyses results of T2D causal genes. (A) The volcano plot revealing 6 potential causal genes for T2D. (B) The box plot showing the expression levels of causal genes for T2D in T2D-PBMCs. (C) The forest plot for the MR results between blood eQTL and T2D. The error bars represent 95%CIs. All statistical tests were two-sided. P < 0.05 was considered significant
Colocalization analyses of candidate genes
Subsequently, GWAS-eQTL colocalization were analyzed (Supplementary Table 3). For the candidate genes, RAP2A (coloc.abf-PPH4 = 0.632) (Fig. 5A-C) supported strong evidence of genetic colocalization, while MCUR1 (coloc.abf-PPH4 = 0.268) (Fig. 5D-F), WNK1 (coloc.abf-PPH4 = 0.302), NFIX (coloc.abf-PPH4 = 0.259) and PANX1 (coloc.abf-PPH4 = 0.372) demonstrated relatively weak colocalization evidence. FOS (coloc.abf-PPH4 = 0.159) presented almost no colocalization evidence (Supplementary Fig. 4).

Colocalization analyses of MCUR1 and RAP2A. (A-C) The regional association plot for colocalization analysis of MCUR1. (D-F) The regional association plot for colocalization analysis of RAP2A
Cell-type specificity expression of causal genes in PBMCs
To explore whether candidate genes had cell type-specific enrichment in PBMCs of patients with periodontitis and T2D, single cell-type expression analysis using single cell RNA-seq (scRNA-seq) data from GEO database was conducted (Fig. 6A). Cells were clustered into 24 clusters and were further classified into 5 cell types (monocyte, NK cell, B cell, T cell, platelet) (Fig. 6B-C). The 6 candidate genes were enriched in different cell types (Fig. 6D; Supplementary Fig. 5). The 2 risk genes, RAP2A and MCUR1 were enriched in NK cells and platelets in PBMCs from patients (Fig. 6E-F).

Single cell-type expression of RAP2A and MCUR1 in PBMCs. (A) The violin plots showing the number of features, RNA counts, percent mitochondrial transcripts and percent hemoglobin found in PBMCs prior to quality control. (B) UMAP representation of 24 cell clusters identified by scRNA-seq. (C) UMAP representation of 5 cell types. (D) Dot plot depicting expression of candidate genes in each cell type. (E) The expression of RAP2A in each cell type. (F) The expression of MCUR1 in each cell type
Discussion
Periodontitis patients have a higher risk of T2D that places substantial socioeconomic burdens on individuals and global health economies [44]. Previous studies focus on the impact of T2D on periodontitis, and this study is the first to leverage a multi-omics integration method to detect the role of periodontitis in T2D pathogenesis and the possible molecular mechanisms. These findings have significant implications for public health and global economy, emphasizing that more attention should be dedicated to periodontal health, as it is a highly modifiable factor that aids in the prevention and management of T2D.
Two-sample MR analysis genetically predicted that periodontitis was causally associated with higher risk of T2D. 79 common DEGs associated with both periodontitis and T2D were then identified in transcriptome data. The integration of GWAS with the eQTLs of these genes from the peripheral blood prioritized 6 genes, including MCUR1, RAP2A, FOS, PANX1, NFIX and WNK1, in the pathogenesis of periodontitis-related T2D. So far, there have been two MR analyses on the relationship between periodontitis and T2D. However, these studies did not exclude all SNPs related to confounders when assessing causality and insufficiently encompassed SNPs to validate causality, reducing the reliability of their results. The potential mechanisms were not further analyzed as well [45, 46]. Our study incorporated the GWAS data from authoritative databases, and the Phenoscanner V2 was used to exclude SNPs associated with confounding factors and outcomes, ensuring highly reliable outcomes [22]. Furthermore, multi-omic approaches and datasets were originally integrated to elucidate the underlying mechanisms by which periodontitis promoted T2D. As verified by our study, colocalization analysis emphasized the importance of RAP2A in the pathogenesis of T2D, and the scRNA-seq datasets indicated that RAP2A was enriched in NK cells, suggesting the potential pathogenic mechanisms.
The peripheral blood tissue is essential for us to characterize genetic effects on gene expression and understand the complicated etiology of periodontitis-related T2D. In the analysis of MR and colocalization, RAP2A was discovered to result in T2D occurrence. RAP2A encodes RAP2A that is a small G protein with GTP-enzyme activity and an important intracellular signal transducer [47]. Only a few studies investigated RAP2A, which mainly focused on the molecular mechanisms of RAP2A in tumor migration and invasion [48, 49]. The current study is the first to investigate the molecular function of RAP2A as a risk factor of T2D, and detect that the rs11065987 was positively associated with T2D risk via increasing the expression of RAP2A according to colocalization analysis. Furthermore, RAP2A may participate in the pathogenesis of periodontitis-related T2D by regulating NK cells, as it was highly expressed in NK cells of patients with two diseases.
MCUR1 is an integral membrane protein that binds to MCU and regulates mitochondrial Ca2+ uptake [50]. Up-regulation of MCUR1 disrupts mitochondrial Ca2+ cycling and has been identified to be associated with several diseases [51,52,53]. Evidence have shown that mitochondrial dysfunction was associated with diabetic periodontitis [54]. Findings from the MR and scRNA-seq of the current study suggested that the evaluated expression of MCUR1 in platelets from PBMCs contributed to the pathogenesis of periodontitis-related T2D, indicating the key roles of mitochondrial dysfunction in promoting T2D. These two risk genes are promising pharmacological targets of periodontitis patients to prevent T2D that merit further exploration.
Another 4 candidates, FOS, PANX1, NFIX and WNK1, were identified from blood tissue and might have protective influence on T2D. However, PANX1, NFIX and WNK1 were up-regulated in T2D patients, which seems to be controversial. Presumably, the up-regulation of PANX1 and WNK1 might be protective response to T2D, as both PANX1 and WNK1 expression positively regulate glucose uptake [55, 56]. Further studies are required to better elucidate the role of NFIX in T2D.
The strength of this investigation is that it used MR and colocalization analyses to estimate the causal effects with the advantages of large sample sizes, minimizing the risk of reverse causation and confounding bias. Multi-omic evidence was also involved to further provide insights into the mechanisms of potential pathogenic genes. Some limitations of this study warrant recognition. Firstly, only 17 SNPs were selected as IVs for periodontitis. The limited number of IVs may introduce bias into the analysis due to collinearity, weak instruments and potential canceling effects among the IVs. Secondly, as the full biological function of the IVs have not been completely elucidated, conclusions cannot be draw that there is no pleiotropy. Despite the use of the MR-Egger method to limit pleiotropy, it has its limitations, such as dependence on the weak instrument assumption, and sensitivity to outliers [28]. As a result, the conclusion drawn from MR-Egger method was not completely reliable. Thirdly, the interpretation of PPH4 in colocalization should be careful, as a low PPH4 may not indicate lacking evidence for colocalization in situations where PPH3 is low as well, which may be the result of limited statistical power [37]. Fourthly, a sex-specific MR analysis on periodontitis and T2D had not been conducted in the present study. Previous studies have indicated a higher prevalence of periodontitis in males compared to females, with the condition often being more severe in males [57, 58], suggesting that males may represent a higher-risk group. Sex-stratified analysis is instrumental in pinpointing subgroups that exhibit varied responses to the exposure, thereby facilitating the implementation of personalized interventions. Ultimately, generalizing the findings of this study to other populations may be inappropriate given its exclusive focus on individuals of European descent.
Conclusions
In conclusion, this study used a multi-omics integration method to explore causality between periodontitis and T2D, and underly molecular mechanisms using multiple bioinformatics tools. MR analyses detected that periodontitis was causally associated with higher risks of T2D and insulin resistance. Furthermore, MCUR1, RAP2A, FOS, PANX1, NFIX and WNK1 were verified to play important roles in the pathogenesis of periodontitis-related T2D, shedding light on the discovery of potential drug targets for periodontitis patients to prevent T2D.
Data availability
The datasets supporting the conclusions of this article are available in the following repositories: DIAGRAM Consortium (http://www.diagram-consortium.org/), the IEU OpenGWAS project (ieu-a-1090) (https://gwas.mrcieu.ac.uk/), FinnGen Release 9 (https://www.finngen.fi/fi), MAGIC (http://magicinvestigators.org/), eQTLGen Consortium (https://eqtlgen.org/), GEO database (GSE189005; GSE10334; GSE244515) (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- T2D:
-
Type 2 diabetes
- MR:
-
Mendelian randomization
- eQTL:
-
Expression quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- DEG:
-
Differentially expressed genes
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PBMC:
-
Peripheral blood mononuclear cell
- GWAS:
-
Genome-wide association study
- IVW:
-
Inverse-variance-weight
- MCUR1:
-
Mitochondrial calcium uniporter regulator 1
- RAP2A:
-
RAS-related protein Rap-2a
- FOS:
-
FBJ murine osteosarcoma viral oncogene homolog
- NFIX:
-
Nuclear factor IX
- PANX1:
-
Pannexin 1
- WNK1:
-
With no lysine-1
References
Collaborators GBDOD, Bernabe E, Marcenes W, Hernandez CR, Bailey J, Abreu LG, Alipour V, Amini S, Arabloo J, Arefi Z, et al. Global, Regional, and national levels and trends in Burden of oral conditions from 1990 to 2017: a systematic analysis for the global burden of Disease 2017 study. J Dent Res. 2020;99(4):362–73.
Chen MX, Zhong YJ, Dong QQ, Wong HM, Wen YF. Global, regional, and national burden of severe periodontitis, 1990–2019: an analysis of the global burden of Disease Study 2019. J Clin Periodontol. 2021;48(9):1165–88.
Slots J. Periodontitis: facts, fallacies and the future. Periodontol 2000 2017, 75(1):7–23.
Cullinan MP, Seymour GJ. Periodontal disease and systemic illness: will the evidence ever be enough? Periodontol 2000. 2013;62(1):271–86.
Molina A, Huck O, Herrera D, Montero E. The association between respiratory diseases and periodontitis: a systematic review and meta-analysis. J Clin Periodontol. 2023;50(6):842–87.
Sanz M, Marco Del Castillo A, Jepsen S, Gonzalez-Juanatey JR, D’Aiuto F, Bouchard P, Chapple I, Dietrich T, Gotsman I, Graziani F, et al. Periodontitis and cardiovascular diseases: Consensus report. J Clin Periodontol. 2020;47(3):268–88.
Jepsen S, Suvan J, Deschner J. The association of periodontal diseases with metabolic syndrome and obesity. Periodontol 2000. 2020;83(1):125–53.
Ahmad E, Lim S, Lamptey R, Webb DR, Davies MJ. Type 2 diabetes. Lancet. 2022;400(10365):1803–20.
Chien WC, Fu E, Chung CH, Cheng CM, Tu HP, Lee WC, Chen WL, Shih KC. Type 2 diabetes Mellitus and Periodontitis: Bidirectional Association in Population-based 15-year Retrospective cohorts. J Clin Endocrinol Metab. 2023;108(11):e1289–97.
Wu CZ, Yuan YH, Liu HH, Li SS, Zhang BW, Chen W, An ZJ, Chen SY, Wu YZ, Han B, et al. Epidemiologic relationship between periodontitis and type 2 diabetes mellitus. BMC Oral Health. 2020;20(1):204.
Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7(12):738–48.
Li W, Peng J, Shang Q, Yang D, Zhao H, Xu H. Periodontitis and the risk of all-cause and cause-specific mortality among US adults with diabetes: a population-based cohort study. J Clin Periodontol 2023.
Shinjo T, Nishimura F. The bidirectional association between diabetes and periodontitis, from basic to clinical. Jpn Dent Sci Rev. 2024;60:15–21.
Tang B, Yan C, Shen X, Li Y. The bidirectional biological interplay between microbiome and viruses in periodontitis and type-2 diabetes mellitus. Front Immunol. 2022;13:885029.
Zhu M, Nikolajczyk BS. Immune cells link obesity-associated type 2 diabetes and periodontitis. J Dent Res. 2014;93(4):346–52.
Lee H, Joo JY, Song JM, Kim HJ, Kim YH, Park HR. Immunological link between periodontitis and type 2 diabetes deciphered by single-cell RNA analysis. Clin Transl Med. 2023;13(12):e1503.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Jansen RC, Nap JP. Genetical genomics: the added value from segregation. Trends Genet. 2001;17(7):388–91.
Mahajan A, Spracklen CN, Zhang W, Ng MCY, Petty LE, Kitajima H, Yu GZ, Rueger S, Speidel L, Kim YJ, et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat Genet. 2022;54(5):560–72.
Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300–10.
Wang Z, Li S, Tan D, Abudourexiti W, Yu Z, Zhang T, Ding C, Gong J. Association between inflammatory bowel disease and periodontitis: a bidirectional two-sample mendelian randomization study. J Clin Periodontol. 2023;50(6):736–43.
Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35(22):4851–3.
Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64.
Yavorska OO, Burgess S. MendelianRandomization: an R package for performing mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46(6):1734–9.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data mendelian randomization. Stat Med. 2017;36(11):1783–802.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Burgess S, Thompson SG. Interpreting findings from mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–89.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some Invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–98.
Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, VanderWeele TJ, Higgins JPT, Timpson NJ, Dimou N, et al. Strengthening the reporting of Observational studies in Epidemiology using mendelian randomization: the STROBE-MR Statement. JAMA. 2021;326(16):1614–21.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–995.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat Genet. 2019;51(5):768–9.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383.
Yuan S, Xu F, Li X, Chen J, Zheng J, Mantzoros CS, Larsson SC. Plasma proteins and onset of type 2 diabetes and diabetic complications: Proteome-wide mendelian randomization and colocalization analyses. Cell Rep Med. 2023;4(9):101174.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–96.
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–72.
Maddux BA, Sbraccia P, Kumakura S, Sasson S, Youngren J, Fisher A, Spencer S, Grupe A, Henzel W, Stewart TA, et al. Membrane glycoprotein PC-1 and insulin resistance in non-insulin-dependent diabetes mellitus. Nature. 1995;373(6513):448–51.
Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365(9467):1333–46.
Chatterjee S, Khunti K, Davies MJ. Type 2 diabetes. Lancet. 2017;389(10085):2239–51.
Shah PD, Schooling CM, Borrell LN. Impact of liability to Periodontitis on Glycemic Control and Type II diabetes risk: a mendelian randomization study. Front Genet. 2021;12:767577.
Wang YB, Yan SY, Li XH, Huang Q, Luo LS, Wang YY, Huang J, Jin YH, Zeng XT. Causal Association between Periodontitis and Type 2 diabetes: a bidirectional two-sample mendelian randomization analysis. Front Genet. 2021;12:792396.
Meng Z, Qiu Y, Lin KC, Kumar A, Placone JK, Fang C, Wang KC, Lu S, Pan M, Hong AW, et al. RAP2 mediates mechanoresponses of the Hippo pathway. Nature. 2018;560(7720):655–60.
Wu JX, Du WQ, Wang XC, Wei LL, Huo FC, Pan YJ, Wu XJ, Pei DS. Rap2a serves as a potential prognostic indicator of renal cell carcinoma and promotes its migration and invasion through up-regulating p-Akt. Sci Rep. 2017;7(1):6623.
Lian Y, Jiang D, Sun J. Tumor suppressive role of miR-33a-5p in pancreatic ductal adenocarcinoma cells by directly targeting RAP2A. Cell Mol Biol Lett. 2021;26(1):24.
Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, et al. MCUR1 is an essential component of mitochondrial Ca2 + uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14(12):1336–43.
Jin M, Wang J, Ji X, Cao H, Zhu J, Chen Y, Yang J, Zhao Z, Ren T, Xing J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):136.
Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, Huang Q, Guo X, Zhang J, Ji L, et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via reactive oxygen species-dependent P53 degradation. Antioxid Redox Signal. 2018;28(12):1120–36.
Garbincius JF, Elrod JW. Mitochondrial calcium exchange in physiology and disease. Physiol Rev. 2022;102(2):893–992.
Sun X, Mao Y, Dai P, Li X, Gu W, Wang H, Wu G, Ma J, Huang S. Mitochondrial dysfunction is involved in the aggravation of periodontitis by diabetes. J Clin Periodontol. 2017;44(5):463–71.
Henriques AFA, Matos P, Carvalho AS, Azkargorta M, Elortza F, Matthiesen R, Jordan P. WNK1 phosphorylation sites in TBC1D1 and TBC1D4 modulate cell surface expression of GLUT1. Arch Biochem Biophys. 2020;679:108223.
Adamson SE, Meher AK, Chiu YH, Sandilos JK, Oberholtzer NP, Walker NN, Hargett SR, Seaman SA, Peirce-Cottler SM, Isakson BE, et al. Pannexin 1 is required for full activation of insulin-stimulated glucose uptake in adipocytes. Mol Metab. 2015;4(9):610–8.
Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. Cdc Periodontal Disease Surveillance workgroup: James Beck GDRP: prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res. 2012;91(10):914–20.
Shiau HJ, Reynolds MA. Sex differences in destructive periodontal disease: a systematic review. J Periodontol. 2010;81(10):1379–89.
Acknowledgements
We thank the FinnGen team, the DIAGRAM Consortium, the IEU OpenGWAS project, the MAGIC, the eQTLGen consortium, and other researchers and participants for providing publicly available data for this analysis.
Funding
This study was supported by the Shuguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (22SG14), Shanghai Young Talents in Healthcare (2022YQ041) and the Shanghai Rising Star Project (22QA1405200).
Author information
Authors and Affiliations
Contributions
X.W., X.Z., contributed to the conception, design, data acquisition, analysis and interpretation, draft and critically revised the manuscript; R.C., Y.L., Y.Y., K.D., contributed to the data acquisition and interpretation, critically revised the manuscript; Z.C., contributed to the data acquisition, critically revised the manuscript; J.S., H.L. contributed to the conception, design, data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The data used in this study have been ethically approved and informed consent has been obtained from the participants in the original research.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wei, X., Zhang, X., Chen, R. et al. Impact of periodontitis on type 2 diabetes: a bioinformatic analysis. BMC Oral Health 24, 635 (2024). https://doi.org/10.1186/s12903-024-04408-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-024-04408-1