
Abstract
Background
ALG3-CDG is a rare autosomal recessive disease. It is characterized by deficiency of alpha-1,3-mannosyltransferase caused by pathogenic variants in the ALG3 gene. Patients manifest with severe neurologic, cardiac, musculoskeletal and ophthalmic phenotype in combination with dysmorphic features, and almost half of them die before or during the neonatal period.
Case presentation
A 23 months-old girl presented with severe developmental delay, epilepsy, cortical atrophy, cerebellar vermis hypoplasia and ocular impairment. Facial dysmorphism, clubfeet and multiple joint contractures were observed already at birth. Transferrin isoelectric focusing revealed a type 1 pattern. Funduscopy showed hypopigmentation and optic disc pallor. Profound retinal ganglion cell loss and inner retinal layer thinning was documented on spectral-domain optical coherence tomography imaging. The presence of optic nerve hypoplasia was also supported by magnetic resonance imaging. A gene panel based next-generation sequencing and subsequent Sanger sequencing identified compound heterozygosity for two novel variants c.116del p.(Pro39Argfs*40) and c.1060 C > T p.(Arg354Cys) in ALG3.
Conclusions
Our study expands the spectrum of pathogenic variants identified in ALG3. Thirty-three variants in 43 subjects with ALG3-CDG have been reported. Literature review shows that visual impairment in ALG3-CDG is most commonly linked to optic nerve hypoplasia.
Similar content being viewed by others


Background
Congenital disorders of glycosylation (CDG) are a rapidly expanding group of genetic multisystem diseases characterized by hypoglycosylation of glycoproteins and glycolipids. Based on the type of glycosidic bond, N- and O-glycosylation disorders are distinguished as two main subclasses of CDG [1].
ALG3-CDG (MIM #601,110) is a rare autosomal recessive disorder of protein N-glycosylation caused by the deficiency of alpha-1,3-mannosyltransferase, which adds one mannose residue in an alpha-1,3 linkage to Man5GlcNAc2-PP-Dol [2].
ALG3-CDG patients show various combinations of cardiac defects (obstructive cardiomyopathy, dilatation of aorta), lung hypoplasia, hepatomegaly, neurological impairment (cerebral and cerebellar atrophy, agenesis of corpus callosum, epilepsy) with developmental delay, musculoskeletal involvement (ulnar deviation, joint contractures, skeletal dysplasia, scoliosis), urogenital abnormalities (nephrocalcinosis, renal cysts, hydronephrosis), microcephaly, visual impairment and dysmorphic features (down slanting palpebral fissures, hypertelorism, high nasal bridge, anteverted nares, large and thick low-set ears with abnormal pinnae, micrognathia, thin lips, inverted nipples). Forty-two cases of ALG3-CDG have been reported to date [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18].
In this study, we characterize in detail the first Czech patient with ALG3-CDG and provide a review of the literature on the identified disease-causing variants and reported ocular findings.
Case presentation
The study was approved by the Ethics committee of the General University Hospital in Prague and adhered to the tenets of the Helsinki Declaration.
The patient was born at 34 6/7 weeks of gestation, with a birth weight of 2.190 kg (34th percentile, -0.41 SD), length of 45 cm (46th percentile, -0.1 SD) and head circumference of 30 cm (16th percentile, -1 SD), following a pregnancy with polyhydramnion and discrete pericardial effusion. She is the first child of non-consanguineous Czech parents. Her early postnatal adaptation was uneventful (Apgar score 8-9-9), but already at birth multiple joint contractures and craniofacial dysmorphic features were noticed. The otoacoustic emissions were absent. Abdominal ultrasound documented multiple renal cysts.
At 3 months of age, the girl was admitted to the hospital with recurrent apnoeic episodes, congenital laryngeal stridor and micro-aspirations. Oral feeding was found to be risky and gastrostomy was placed. On clinical examination she presented with central hypotonia, plagiocephaly, abnormal subcutaneous fat distribution around the neck, port-wine stain on the forehead and craniofacial dysmorphia (Fig. 1A, 1B). Length (55 cm, 12th percentile) and weight (4.66 kg, 39th percentile) were normal but there was severe progressive microcephaly (head circumference 34.5 cm, <1st percentile, -3.49 SD). Laboratory investigations showed antithrombin (35 %, controls 80–140 %) and factor XI deficiency (42 %, controls 55–135 %).

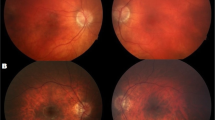
Clinical findings in the Czech case with ALG3-CDG. Photograph of the girl at the age of 4 months (A, B) and at the age of 23 months (C-E). Note microcephaly, port-wine stain on the forehead, hypertelorism, wide and flattened nasal bridge, long and smooth philtrum, thin upper lip (A), dysplastic and low set ears with malformed pinnae (B), down-slanting palpebral fissures, mild ptosis and anteverted nares, contracted wrists and long fingers, semiflexed knees, gastrostomy (C). Detailed photographs of the hand and leg showing flexed joints, long fingers with ulnar deviation, adducted thumbs (D), rigid clubfoot, medial protrusion of the navicular bone, lateral deviation of high arch and toes (E). Wide-field fundus photograph of the right eye taken at the age of 23 months showing optic nerve hypoplasia and hypopigmentation (F). SD-OCT horizontal perimacular scan of the left eye (G), note inner retinal layer thinning together with preservation of the external limiting membrane, ellipsoid zone and interdigitation zone. Analysis of the ganglion cell layer using automatic segmentation documents profound loss (H) when compared to a scan obtained from a similarly aged healthy female child (I). The quality of retinal imaging was limited by compliance, images of the left eye could not be obtained
Neurological examination showed spastic tetraplegia, hyporeflexia and developmental delay. Although clinical seizures were not noticed, electroencephalography (EEG) was severely abnormal showing high amplitude slow spike-wave complexes (SSWC) and slow background activity. Anticonvulsant therapy was therefore initiated. The brain magnetic resonance imagining showed cerebral and cerebellar atrophy, cavum septum pellucidum, enlarged 4th ventricle and cisterna magna, delayed myelinization, and optic nerve hypoplasia.
Ophthalmic examination showed hypopigmentation of the fundus and pale optic discs. Visual and brain stem auditory evoked potentials (VEP, BAEP) were both pathological with prolonged latencies of wave N70 and waves III, V, respectively. Cardiological examination was normal apart from insignificant patent arterial duct and patent foramen ovale.
At 12 months of age, epilepsy was not compensated despite combined therapy with levetiracetam and phenobarbital. Add-on therapy with topamirate was started. At that time, although the development was severely delayed, the patient began to fix objects and to react to sounds and voices of family members, gurgle and lift up her head.
Despite gastrostomy feeding, all her anthropometric parameters severely declined with length 66 cm (< 1st percentile, -2.35 SD), weight 7.38 kg (6th percentile, -1.96 SD) and head circumference 38 cm (< 1st percentile, -5.17 SD). EEG at 14 months showed similar pattern to the earlier recordings with slow background activity and multiple SSWC.
At the last follow-up, the girl was 23 months-old, severe developmental delay was apparent, seizures were ameliorated on triple anticonvulsant medication. Her recurrent micro-aspirations were life threatening. She showed further deceleration of the anthropometric data: length 77 cm (< 1st percentile, -2.69 SD), weight 10.2 kg (6th percentile, -1.96 SD) and head circumference 40 cm (< 1st percentile, -5.53 SD). On physical examination, flexed joints, long fingers with ulnar deviation, adducted thumbs and laterally deviated toes were observed (Fig. 1 C-1E).
We performed esophagogastroduodenoscopy that revealed esophagitis and duodenum ulcer. Repeated abdominal ultrasonography documented multiple cysts in both kidneys which were of stable size since birth, other abdominal organs were without abnormalities.
Ocular examination including spectral-domain optical coherence tomography (SD-OCT) (Spectralis, Heidelberg Engineering GmbH, Heidelberg, Germany) revealed profound loss of retinal ganglion cells and inner retinal layer thinning with preservation of the external limiting membrane, ellipsoid zone and interdigitation zone (Fig. 1 F-1I). Fundus appeared hypopigmented and optic discs were pale as documented using ultra-wide field camera (Clarus 700, Carl Zeiss Meditec AG, Jena, Germany). Visual activity could not be measured.
Transferrin isoelectric focusing was performed as described [19] and showed a type 1 pattern.
DNA from the proband and her parents was extracted from peripheral leucocytes. Targeted sequencing of 260 genes related to neuromuscular diseases and arthrogryposis was performed by hybrid capture enrichment (Roche NimbleGen, Pleasonton, CA, USA) and massive parallel sequencing (NextSeq, Illumina, San Diego, CA, USA) as described [20]. Sequencing data were analysed with Sequence Pilot software (JSI medical systems, Ettenheim, Germany). Conventional Sanger sequencing was used to verify the presence of presumed causal variants and for targeted screening in first-degree relatives. The effect of one missense mutation was predicted using five computational tools: Sorting Intolerant from Tolerant (SIFT) [21], Polyphen-2 [22], Mutation Taster2 [23], M-CAP [24] and CADD [25]. The pathogenicity of the detected variants was evaluated according to the American College of Medical Genetics and Genomics (ACMG) recommendations [26].
Two ALG3 variants (NM_005787.6) c.116del p.(Pro39Argfs*40) and c.1060 C > T p.(Arg354Cys) were identified, neither of them has been reported. Segregation analysis confirmed their trans position, the c.116del was inherited from the mother while the c.1060 C > T from the father. All five prediction tools used evaluated the missense variant as deleterious or likely deleterious. Based on the available evidence both variants were classified as pathogenic according to the ACMG criteria.
Discussion
In this study we describe clinical, biochemical, and molecular genetic findings of an unreported ALG3-CDG patient. We also review ocular and molecular genetic findings in all 43 individuals with ALG3-CDG identified to date.
Our case adds to the previously published phenotype data several novel aspects. In earlier reports pale or small optic disc was observed in 12 patients suggesting optic nerve atrophy or hypoplasia [2, 5, 6, 8, 9, 11, 14, 18]. Our case report is however the first clearly documenting optic nerve and retinal layers pathology by ocular imaging methods.
Clinical course
Out of the 43 reported individuals 17 had a lethal phenotype with prenatal manifestation, medical termination of pregnancy and/or early neonatal death. Three further patients died of respiratory failure during pneumonia in childhood (aged 1.8; 3.5 and 6 years) [11, 12, 14] and one of multiorgan failure (aged 1 year) [18]. Neurological symptoms have recently been summarized in 26 patients, and a ketogenic diet was suggested as a therapeutic option for intracrable epilepsy [16]. Dysmorphic and musculoskeletal features have been also recently reviewed in 19 patients [15].
The longest living patient is 38 years-old male suffering from moderate intellectual disability, intractable seizures, feeding difficulties, facial dysmorphism, hearing loss, cardiac abnormalities, osteopenia/osteoporosis, nocturnal apnoea, scoliosis, hypothyroidism [18].
Ocular phenotype
Presumably because of poor compliance due to young age at examination and developmental delay available ophthalmic data in ALG3-CDG are limited (Table 1). Most often poor eye fixation, strabismus and/or nystagmus have been noted. However, the likely underlying cause was only reported in few patients and comprised optic nerve atrophy/hypoplasia observed in 11 patients including the current study confirming deficiency of retinal ganglion cells by SD-OCT, and signs of chorioretinal dystrophy in three patients, of these one had also optic atrophy [11]. Electroretinography was reported in three patients; decreased amplitudes of both rod and cone responses were observed [4, 7].
Less common ocular features of ALG3-CDG were congenital cataract found in one individual [10] and iris coloboma present in another patient [2]. One fetus examined after pregnancy termination at week 25 was noted to have proptosis and corneal opacities [10]. Another fetus terminated in week 28 had eyelid ptosis [15].
Disease-causing variants
Sequence variants previously reported as causing ALG3-CDG (reference sequence NM 005787.6) were searched in the literature. The population frequency of the variants was retrieved from the Genome Aggregation Database v2.1.1 from 125,748 human exomes and 15,708 genomes [27]. In total 33 pathogenic/likely pathogenic variants in ALG3 have been found in 43 individuals with ALG3-CDG from 29 families (Table 2). Most subjects carried a unique disease-causing variant(s), only three mutations have been identified recurrently. Homozygous variants were mainly linked to consanguinity when this information was available [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18].
Conclusions
CDG pose a diagnostic challenge due to their high phenotypic variability. Combination of severe neurologic and visual impairment with dysmorphia, arthrogryposis and other congenital malformations should raise suspicion of a CDG syndrome.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- ALG3:
-
Asparagine-linked glycosylation 3
- BAEP:
-
Brain stem auditory evoked potentials
- CDG:
-
Congenital disorders of glycosylation
- gnomAD:
-
Genome aggregation database
- EEG:
-
Electroencephalography
- MIM:
-
Mendelian Inheritance in Man
- SD:
-
Standard deviation
- SD-OCT:
-
Spectral-domain optical coherence tomography
- SSWC:
-
Slow spike-wave complexes
- VEP:
-
Visual evoked potentials
References
Hennet T: Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim Biophys Acta 2012, 1820(9):1306–1317.
Korner C, Knauer R, Stephani U, Marquardt T, Lehle L, von Figura K: Carbohydrate deficient glycoprotein syndrome type IV: deficiency of dolichyl-P-Man:Man(5)GlcNAc(2)-PP-dolichyl mannosyltransferase. EMBO J 1999, 18(23):6816–6822.
Denecke J, Kranz C, Kemming D, Koch HG, Marquardt T: An activated 5’ cryptic splice site in the human ALG3 gene generates a premature termination codon insensitive to nonsense-mediated mRNA decay in a new case of congenital disorder of glycosylation type Id (CDG-Id). Hum Mutat 2004, 23(5):477–486.
Denecke J, Kranz C, von Kleist-Retzow J, Bosse K, Herkenrath P, Debus O, Harms E, Marquardt T: Congenital disorder of glycosylation type Id: clinical phenotype, molecular analysis, prenatal diagnosis, and glycosylation of fetal proteins. Pediatr Res 2005, 58(2):248–253.
Schollen E, Grunewald S, Keldermans L, Albrecht B, Korner C, Matthijs G: CDG-Id caused by homozygosity for an ALG3 mutation due to segmental maternal isodisomy UPD3(q21.3-qter). Eur J Med Genet 2005, 48(2):153–158.
Sun L, Eklund EA, Chung WK, Wang C, Cohen J, Freeze HH: Congenital disorder of glycosylation id presenting with hyperinsulinemic hypoglycemia and islet cell hyperplasia. J Clin Endocrinol Metab 2005, 90(7):4371–4375.
Kranz C, Sun L, Eklund EA, Krasnewich D, Casey JR, Freeze HH: CDG-Id in two siblings with partially different phenotypes. Am J Med Genet A 2007, 143A(13):1414–1420.
Rimella-Le-Huu A, Henry H, Kern I, Hanquinet S, Roulet-Perez E, Newman CJ, Superti-Furga A, Bonafe L, Ballhausen D: Congenital disorder of glycosylation type Id (CDG Id): phenotypic, biochemical and molecular characterization of a new patient. J Inherit Metab Dis 2008, 31 Suppl 2:S381-386.
Riess S, Reddihough DS, Howell KB, Dagia C, Jaeken J, Matthijs G, Yaplito-Lee J: ALG3-CDG (CDG-Id): clinical, biochemical and molecular findings in two siblings. Mol Genet Metab 2013, 110(1–2):170–175.
Lepais L, Cheillan D, Frachon SC, Hays S, Matthijs G, Panagiotakaki E, Abel C, Edery P, Rossi M: ALG3-CDG: Report of two siblings with antenatal features carrying homozygous p.Gly96Arg mutation. Am J Med Genet A 2015, 167A(11):2748–2754.
Fiumara A, Barone R, Del Campo G, Striano P, Jaeken J. Electroclinical features of early-onset epileptic encephalopathies in congenital disorders of glycosylation (CDGs). JIMD Rep. 2016, 27:93–99.
Barba C, Darra F, Cusmai R, Procopio E, Dionisi Vici C, Keldermans L, Vuillaumier-Barrot S, Lefeber DJ, Guerrini R, Group CDG: Congenital disorders of glycosylation presenting as epileptic encephalopathy with migrating partial seizures in infancy. Dev Med Child Neurol 2016, 58(10):1085–1091.
Alsubhi S, Alhashem A, Faqeih E, Alfadhel M, Alfaifi A, Altuwaijri W, Alsahli S, Aldhalaan H, Alkuraya FS, Hundallah K et al: Congenital disorders of glycosylation: The Saudi experience. Am J Med Genet A 2017, 173(10):2614–2621.
Himmelreich N, Dimitrov B, Geiger V, Zielonka M, Hutter AM, Beedgen L, Hullen A, Breuer M, Peters V, Thiemann KC et al: Novel variants and clinical symptoms in four new ALG3-CDG patients, review of the literature, and identification of AAGRP-ALG3 as a novel ALG3 variant with alanine and glycine-rich N-terminus. Hum Mutat 2019, 40(7):938–951.
Bian Y, Qiao C, Zheng S, Qiu H, Li H, Zhang Z, Yin S, Jiang H, Li-Ling J, Liu C et al: ALG3-CDG: lethal phenotype and novel variants in Chinese siblings. J Hum Genet 2020, 65(12):1129–1134.
Paketci C, Edem P, Hiz S, Sonmezler E, Soydemir D, Sarikaya Uzan G, Oktay Y, O’Heir E, Beltran S, Laurie S et al: Successful treatment of intractable epilepsy with ketogenic diet therapy in twins with ALG3-CDG. Brain Dev 2020, 42(7):539–545.
Ferrer A, Starosta RT, Ranatunga W, Ungar D, Kozicz T, Klee E, Rust LM, Wick M, Morava E: Fetal glycosylation defect due to ALG3 and COG5 variants detected via amniocentesis: Complex glycosylation defect with embryonic lethal phenotype. Mol Genet Metab 2020, 131(4):424–429.
Alsharhan H, Ng BG, Daniel EJP, Friedman J, Pivnick EK, Al-Hashem A, Faqeih EA, Liu P, Engelhardt NM, Keller KN et al: Expanding the phenotype, genotype and biochemical knowledge of ALG3-CDG. J Inherit Metab Dis 2021.
Guillard M, Wada Y, Hansikova H, Yuasa I, Vesela K, Ondruskova N, Kadoya M, Janssen A, Van den Heuvel LP, Morava E et al: Transferrin mutations at the glycosylation site complicate diagnosis of congenital disorders of glycosylation type I. J Inherit Metab Dis 2011, 34(4):901–906.
Stehlikova K, Skalova D, Zidkova J, Haberlova J, Vohanka S, Mazanec R, Mrazova L, Vondracek P, Oslejskova H, Zamecnik J et al: Muscular dystrophies and myopathies: the spectrum of mutated genes in the Czech Republic. Clin Genet 2017, 91(3):463–469.
Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC: SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012, 40(Web Server issue):W452-457.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR: A method and server for predicting damaging missense mutations. Nat Methods 2010, 7(4):248–249.
Schwarz JM, Cooper DN, Schuelke M, Seelow D: MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014, 11(4):361–362.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G: M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016, 48(12):1581–1586.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M: CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019, 47(D1):D886-D894.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17(5):405–424.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP et al: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581(7809):434–443.
Acknowledgements
None.
Funding
This work was supported by RVO-VFN 64165 project of the Ministry of Health of the Czech Republic, further funding was provided by UNCE/MED/007, SVV 260516 and PROGRES-Q26/LF1 programs of the Charles University. TH, HH and NO were supported by EUROGLYCAN-omics, Ministry of Education Youth and Sports of Czech Republic No. 8F19002, under the frame of E-Rare-3, the ERA-Net for Research on Rare Diseases; PL, TH, BK and AČ by AZV NU20-07-00182 research grant from the Ministry of Health of the Czech Republic.
Author information
Authors and Affiliations
Contributions
MF and AC conducted the review and wrote a substantial part of the manuscript including the figure and tables. TH, MF, BK, and PL examined the patient. TH and PL contributed to the manuscript with advice and critical comments, supervised the manuscript preparation. HH and NO performed the biochemical analyses. JZ did the molecular genetic study. The manuscript was reviewed and revised by all the authors. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics committee of the General University Hospital in Prague (reference no. 34/19; date 23.5.2019). Written informed consent was obtained from the patient’s parents before inclusion.
Consent for publication
We obtained from the patient’s parents written informed consent for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Farolfi, M., Cechova, A., Ondruskova, N. et al. ALG3-CDG: a patient with novel variants and review of the genetic and ophthalmic findings. BMC Ophthalmol 21, 249 (2021). https://doi.org/10.1186/s12886-021-02013-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-021-02013-2