
Abstract
Background
Rectal mucosal melanoma (RMM) is a rare and highly aggressive disease with a poor prognosis. Due to the rarity of RMM, there are few studies focusing on its genetic mechanism. This retrospective study aimed to analyze the genetic spectrum and prognosis of RMM in China and lay a foundation for targeted therapy.
Methods
36 patients with primary RMM from Peking University Cancer Hospital were enrolled in this study. The Next-generation sequencing (NGS) data of the tumor samples were fitted into the TruSight™ Oncology 500 (TSO500) Docker pipeline to detect genomic variants. Then, the univariate and multivariate Cox hazard analysis were performed to evaluate the correlations of the variants with the overall survival (OS), along with Kaplan-Meier and log-rank test to determine their significance.
Results
BRAF mutations, NRG1 deletions and mitotic index were significant prognostic factors in the univariate analysis. In multivariable analysis of the OS-related prognostic factors in primary RMM patients, it revealed 2 significant alterations: BRAF mutations [HR 7.732 (95%CI: 1.735–34.456), P = 0.007] and NRG1 deletions [HR 14.976 (95%CI: 2.305–97.300), P = 0.005].
Conclusions
This is the first study to show genetic alterations exclusively to Chinese patients with RMM. We confirmed genetic alterations of RMM differ from cutaneous melanoma (CM). Our study indicates that BRAF and NRG1 were correlated with a poor prognostic of RMM and may be potential therapeutic targets for RMM treatment.
Similar content being viewed by others


Introduction
Mucosal melanoma (MM) is a malignant tumor caused by the aberrant growth of melanocytes in the mucosal lining. As a subtype of melanoma, MM may occur in the mucosal layer of different anatomical parts, such as the respiratory tract, gastrointestinal tract and urogenital tract. Compared with cutaneous melanoma (CM), MM is particularly rare. In the United States, MM accounts for only 1.3% of melanoma, while melanoma originated from skin accounts for 91.2% [1]. However, in a study enrolling 522 Chinese melanoma patients, the incidence of MM was 22.6% [2]. Anorectal mucosal melanoma (ARMM) is one of the most common types of MM, often accompanied by a less favorable prognosis [3]. Although melanoma occurring in rectum is more common than that in anus, most studies always combine these two entities [4, 5]. In addition, Tchelebi et al. showed that the incidence of rectal mucosal melanoma (RMM) continued to rise, with a higher rate of growth than anal melanoma over the past decades [6]. However, whether the lesion is located in anus or rectum, the long-term prognosis is poor [7]. Due to the low incidence of the disease and less reported, the etiology, pathogenesis and genetic cognition of RMM are still unclear, which brings difficulties to the correct diagnosis, treatment and prognosis prediction.
So far, surgery has been recommended as the optimal treatment for this rare tumor, allowing for complete local excision [8, 9]. The additional radiotherapy does not improve prognosis of patients with RMM, even those with more locally advanced disease [6]. Chemotherapy and biochemotherapy showed limited efficacy on MM patients, while there was no significant survival improvement in CM patients [10]. Despite surgical resection and emergence of various forms of adjuvant therapy, the overall prognosis of RMM remains dismal [11]. For CM, targeted therapy and immunotherapy have been shown to improve prognosis in advanced patients [12]. However, at present, the applicability of targeted treatment based on BRAF inhibitors is limited because the incidence of BRAF mutations in MM is much lower than that in CM [13]. Therefore, the discovery of potential therapeutic targets of RMM is particularly important.
It is well documented that mutational profiles of malignant melanomas in China are significantly different from that in Western countries [14, 15]. A genome-wide landscape study also confirmed that MM showed different mutation characteristics in different anatomical parts [16]. Therefore, we analyzed genetic alterations of 36 Chinese primary RMM verifying the difference of alterations between MM and CM. In addition, we revealed that BRAF and NRG1 could be potential therapeutic targets for RMM. Our study depicted the mutation landscape of Chinese RMM and provides more insights into its targeted treatment.
Method
Patients and pathology
We enrolled 36 RMM patients treated in Peking University Cancer Hospital from May 2010 to March 2019, along with their clinical and sequencing data. This retrospective study was approved by the ethics committee of Peking University Cancer Hospital. All patients have signed the informed consent. Clinical data included age at diagnosis, sex, treatment and follow-up results. All cases have formalin-fixed and paraffin-embedded (FFPE) samples available for analyses and they were confirmed diagnosed by 2 experienced pathologists in the pathology department. The following primary tumor clinicopathological characteristics were assessed: depth of tumor invasion (lamina propria, submucosa, muscularis propria or serosa and beyond); pigment content; thickness; vascular invasion; perineural invasion; ulcer of primary focus; mitotic index and lymph node metastasis (Table 1).
Immunohistochemistry (IHC) staining
Common pathological markers of melanoma including CK, SOX-10, Melan-A, HMB-45 and S-100 were identified by IHC staining. The IHC results were independently evaluated by two different pathologists, who did not know the clinical information related to the subjects. If the two pathologists got different results, a third pathologist would review the slide. For SOX-10, IHC staining of any tumor cell nucleus was positive; for HMB-45, Melan-A and S-100, IHC staining was positive when any tumor cells showed cytoplasmic staining, and for CK, IHC staining was negative when all tumor cells showed loss of membrane staining. The tumor was considered to be MM if at least one staining of Melan-A, HMB-45, S-100, and SOX-10 was positive and CK was negative.
DNA and RNA extractions
Genomic DNA and RNA samples were extracted from the same FFPE tissue sections using kit AllPrep DNA/RNA FFPE KIT (Cat No./ID:80234) according to the manufacturer’s protocols.
In this study, the preparation of both RNA and DNA sample library were performed according to the TSO500 Library Preparation Kit (Illumina, San Diego, CA, USA). Then, a two-step capture and enrichment of specific capture probes for DNA samples was performed, the DNA library and corresponding cDNA library of 8 samples was standardized by using the library homogenization method based on magnetic bead purification and sequenced using the Illumina NextSeq 550Dx platform. The Sequencing results were analyzed using TSO500 Docker pipeline.
Sequence data processing
The sequence alignment to the human genome (hg19) was completed using the BWA-MEM (version 0.7.11) [17] alignment algorithm. SAMtools (version 1.3) [18] was used to do bam-sam conversions. We used Genome Analysis Toolkit (GATK, version 3.6) [19] module IndelRealigner was used to perform local realignment of indels. Germline variants were filtered out using in-house built database, and all parameter were set according to [20]. Copy number variants (CNVs) including amplification and deletion were identified by CRAFT copy-number callers from the TSO500 pipeline. Manta (version 1.6.0) [21] was employed to detect large-scale structure variations (SVs) in RNA library, and only fusions having at least 3 unique supporting reads, one of which being a split read crossing the fusion breakpoint, are considered as candidate fusions.
Measurement of immunotherapy biomarkers
Tumor mutation burden (TMB) was measured as the number of eligible somatic mutations (coding or high-confidence regions, over 50x coverage, over 5% VAF single-nucleotide variants (SNVs) and indels) by NGS per Megabase (Mb), after filtering germline variants (in-house database) and high COSMIC database counts mutations. Only the SNVs and indels in the coding regions were considered for TMB measurement. Assessment of raw sequencing data and TMB were conducted with the Illumina TSO500 supporting software. TMB-High and TMB-Low were divided by the median TMB score of the 36 RMM patients.
Samples’ microsatellite status was determined using TSO500 Docker pipeline in the form of microsatellite instability (MSI) score. Samples with MSI score ≥ 20 were considered as MSI, otherwise microsatellite stability (MSS).
Statistical analysis
The primary endpoint of our study was overall survival (OS). OS is defined as the time interval between the date of tumor resection and date of death from any cause or the date of the last follow-up. SPSS software (SPSS, version 20.0) was used to perform statistical analysis. The OS rate was described using Kaplan-Meier survival curves and log-rank test. The Cox proportional hazards model was used to perform univariate and multivariate analysis. And the hazard ratio and 95% confidence interval (95%CI) were recorded for parameters. The statistical significance level was set at 0.05.
Result
Clinicopathological characteristics
A total of 36 patients with RMM were included in this study. Our clinicopathological analyze was predominantly based on the primary cases. The male/female ratio was 1:1.57, and the median age was 62 years (range, 43–98 years). Other distribution of relevant parameters of 36 primary RMM patients was summarized in Table 1.
A majority of RMM patients had lymph node metastasis (61.1%), while 33.3% RMM did not occur and 5.6% remained undetermined. As RMM is not included in the American Joint Committee on Cancer (AJCC) melanoma staging system, we used the depth of tumor invasion which is similar to the T staging used in colorectal cancer. The level of invasion for the non-perianal tumors was either submucosal (10/36, 27.8%), to muscularis propria (20/36, 55.6%) and beyond the muscularis propria into perirectal soft tissue (6/36, 16.6%). It is worth noting that most of patients presented with ulcers (83.3%). Vascular and perineural invasion were identified in 17 (47.2%) and 11 (30.6%) tumors, respectively. The median thickness of the tumor is 11 mm.
Mutation profile of RMM
We performed NGS on tumor samples from 36 RMM patients and assessed the SNVs and the CNVs. There were 183 gene mutations found in 36 RMM samples, with an average TMB of 9.15 mut/Mb and all patients were assessed as MSS.
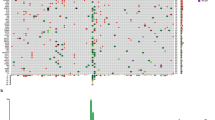
All the mutant genes were listed in Table S1, while Fig. 1 depicted the mutation profile of top 35 genes. Our results showed that KIT was the most frequently mutated gene (33%) in RMM, and there were 5 patients with KIT mutation at site p.L576P, one of which had concomitant mutation at site p.W557R. In addition, BRAF mutations were diverse, with only one p.V600E mutation and the remaining three mutations were p.D594G, p.G469A, p.R146Q. SF3B1 were identified in 5 patients (13.9%), 3 of whom showed hotspot mutation in R625H. Other melanoma-related genes were also discovered in RMM mutations, including TP53 (13.8%) and NRAS (2.8%). Besides, genes with moderate mutation frequency were observed, such as SPTA1, BCR, MGA, GNAS and BARD1 (Figure S1).

Mutation landscape of 36 RMM (top 35). All somtatic mutations or CNVs that occurred in 3 or more samples were depicted in the figure, along with TMB and clinical information
The top 8 frequent CNVs were depicted in Fig. 1, and more details were in Table S2. In our study, MYC (9/36, 25.0%), CCND3 (7/36, 19.4%) and PDGFRA (8/36, 22.2%) showed the most frequent amplification/deletion rates. KIT amplifications were detected in 19.4% of cases, mostly on chromosome 4. Interestingly, KIT p.L576P mutation existed simultaneously with amplification in one case. BRAF amplification was detected in two cases. In addition, we also observed copy number deletions, including PTEN (16.7%) and NRG1 (8.3%). Our data suggested that there were also gene fusions in RMM patients, where one NTRK gene fusion and one BRAF gene fusion were identified.
Identification of OS-related markers
We analyzed the association between patients’ OS with clinicopathological characteristics. Of all 36 patients, 12 were alive at the end of follow-up. Prognosis was poor for all patients, with median OS of only 15.7 months (25–75% quartiles: 11.3 months – 40.8 months). Mitotic index (the median of mitotic index served as the threshold value for grouping) and lymph node metastasis were significant in univariate analysis related to OS (P = 0.044 and P = 0.045, respectively), while other clinicopathological characteristic showed no significant differences (Table S3).
In order to evaluate the relationship between gene alterations and the survival of MM patients, a gene with mutation frequency or CNVs frequency greater than 8.0% was selected for survival analysis. As a result, we found that patients with BRAF mutation had significantly shorter OS than non-mutated ones (P = 0.008, Table 2, Fig. 2A). Besides, NRG1 copy number deletion was present in a total of 8.3% of the cohort and its correlation with OS remained significant (P = 0.001, Table 2, Fig. 2B), while other alterations (KIT, SF3B1, NF1 and TP53) did not show the same performance (Table 2).

Kaplan-Meier survival curves of OS. A BRAF mutations was significantly associated with poor OS (P = 0.008). B NRG1 CNVs was significantly associated with poor OS (P = 0.001)
The multivariate model was employed to evaluate the comprehensive prognostic value of features obtained from univariate analysis, as shown in Table 3. It revealed 3 significant prognosis-related variables: lymph node metastasis, BRAF mutations and NRG1 deletions, where the significance level for lymph node metastasis was much close to 0.05, and limited to the number of samples, we did not perform an in-depth analysis on it. The overview of our analysis was demonstrated in Figure S2.
Discussion
In a retrospective cohort involving 446 Chinese patients with melanoma, melanoma was shown to be more aggressive and had a shorter OS than cutaneous lesions [22]. Although similar research has been reported in previous studies, no comprehensive analysis of specific mucosal site has been carried out [15, 23, 24]. In this study, NGS technology was applied to depict the mutation spectrum of 36 RMM patients to provide a reference for the clinical prognosis and further targeted intervention therapy.
A review of the histopathology revealed multiple adverse prognostic pathological factors including deep tumor thickness, ulceration, high tumor mitotic rate and lymphovascular invasion [25]. In our study, univariate analysis confirmed that mitotic index and lymph node metastasis may be risk factors of prognostic significance. Apart from this, Nagarajan et al. found the presence of lymphovascular invasion was correlated with shorter disease-specific survival in ARMM [26], which was also consistent with our research.
Similar to the previous findings [15, 27,28,29], we identified recognized mutations associated with MAPK signaling pathway, including mutations in NF1, KIT, NRAS and BRAF. This provides a reliable theory for targeted therapy of RMM. In the meantime, the mutation profile also suggested that RMM’s mutation characteristic may be different from that of CM [23]. It is well known that BRAF mutations occur in more than 50% of CM, and targeted therapy for melanoma has been applied clinically which achieved good efficacy. However, BRAF mutations in MM are less frequent than that in CM. A review about BRAF mutations collected in 1339 cases of mucosal melanoma revealed that BRAF mutations were present in 8.0% (107/1339) MM [13]. In two studies focusing on MM molecular spectrum, the frequency of BRAF mutations were reported as 3.1% (2/65) [30] and 7.5% (3/40) [31], respectively. These results are similar to ours (11.1%, 4/36). Moreover, there are significant differences between CM and MM in the location of BRAF mutations. It has been documented that small molecule inhibitors (vemurafenib and dabrafenib) of BRAF V600–mutant induce tumor regression, and combination of BRAF and MEK inhibitors can improve survival rate of melanoma patients [32,33,34].
Although BRAF mutations are rare in MM, they are characterized by a higher prevalence of non-V600 mutations than in CM [13], which was also confirmed in our study. It is notable that although the cohort was small, the SNV of BRAF was associated with the poor prognosis. Currently, only few studies have investigated the molecular mechanisms of non-BRAF V600 mutations, which are able to promote MEK phosphorylation in a CRAF-dependent manner by directly binding to and activating CRAF to drive the MAPK pathway [35]. In consistent with the report of genetic alterations in ARMM [26, 28, 36], other genes such as SF3B1 and TP53, also mutated in our cohort. Apart from these common mutations, many genes had intermediate mutation frequencies in RMM, including BARD1, SPTA1, MGA, and BCR. These genes are associated with the occurrence of other malignancies, but have not been reported in MM, indicating that the potential therapeutic targets for RMM needs to be further studied.
Our results revealed that most RMM patients have CNVs mutations, and MYC was most frequently amplified, which was consistent with previous melanoma studies [31, 37,38,39,40]. High MYC expression is often associated with tumor metastasis and poor prognosis in melanoma [41]. However, we did not observe a significant difference of OS between MYC amplification and MYC non-amplification cohorts. In addition, PDGFRA, CCND3, KIT amplification and PTEN deletion have been reported in previous studies of MM [30, 38], which was consistent with our findings. Deletion of the NRG1 gene was detected in 8.3% (3/36) of RMM patients and was associated with poor prognosis in our cohort. Neuregulins (NRGs) are a large subclass of polypeptide growth factors of the epidermal growth factor (EGF) family [42]. On one hand, it can specifically bind to the extracellular domain the receptor tyrosine kinase ERBB3 and ERBB4 to alter receptor conformation and promote dimerization with ERBB2 [42]. Receptor hetero dimerization promotes autophosphorylation of the cytoplasmic tyrosine residues, resulting in the activation of downstream PI3 kinase and MAP kinase signaling pathways [43]. NRG1/ERBB3 signaling was able to negatively regulate melanocyte (MC) differentiation and pigmentation while promoting proliferation [43]. On the other hand, the NRG1 gene is frequently silenced by methylation in breast cancers and NRG1 may be the principal tumor suppressor gene that leads to loss of the short arm of chromosome 8 in many breast and other epithelial cancers [44]. The NRG1 gene has been proposed both as a candidate oncogene and a candidate tumor suppressor gene [44]. Previous studies have shown that over expression of NRG1 leads to the activation of ERBB3/ERBB2 signaling and a poor prognosis. One of the reasons might be the paracrine effect of NRG1 enhances the resistance to RAF and MEK inhibitors [45, 46]. However, no deletion alteration of NRG1 has been reported in RMM previously. Further studies are necessary to explore the biological significance of NRG1 deletion in RMM patients and how this affects the progression of melanoma. BRAF amplifications were also observed in this study. It is worth noting that BRAF amplification resulting in BRAF over-expression has been reported as one of the mechanisms responsible for acquired resistance to BRAF and/or MEK inhibitor [47, 48]. As has been documented, some common genetic variants found in MM, such as amplifications of CDK4, MDM2 and TERT or deletions of CDKN2A and ATM, were rare in our RMM cohort [15, 30]. Therefore, these results may reveal a unique pattern of CNVs in RMM patients.
Rare gene fusions that have been reported in MM was also observed in our cohort. NTRK gene fusion has been reported in colonic MM [49] and could be a novel target of NTRK inhibitors [50]. Kim et al. identified a novel ZNF767-BRAF gene fusion that showed resistance to the BRAF inhibitor vemurafenib in respiratory MM patients [51]. Although these two fusion genes have been detected in our cohort, their biological mechanism remains unclear.
Conclusions
To our best knowledge, this is the first study specifically for genetic alterations in Chinese RMM patients. We performed NGS analysis of 36 primary RMM specimens to describe the genetic characteristics and explore the pathogenesis of this rare tumor. Here, we confirmed that the genetic variation of RMM is different from that of CM, and reported the possibility of BRAF and NRG1 as potential targets of RMM.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 95%CI:
-
95% confidence interval
- AJCC:
-
American Joint Committee on Cancer
- ARMM:
-
Anorectal mucosal melanoma
- CM:
-
Cutaneous melanoma
- CNVs:
-
Copy number variants
- EGF:
-
Epidermal growth factor
- FFPE:
-
Formalin-fixed and paraffin-embedded
- HR:
-
Hazard ratios
- IHC:
-
Immunohistochemical
- Mb:
-
Megabase
- MC:
-
Melanocyte
- MM:
-
Mucosal melanoma
- MSI:
-
microsatellite instability
- MSS:
-
Microsatellite stable
- NGS:
-
Next generation sequencing
- NRGs :
-
Neuregulins
- OS:
-
Overall survival
- RMM:
-
Rectal mucosal melanoma
- SNVs:
-
Single-nucleotide variants
- TMB:
-
Tumor mutation burden
- TSO500:
-
TruSight™ Oncology 500
References
Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. Cancer. 1998;83(8):1664–78. https://doi.org/10.1002/(SICI)1097-0142(19981015)83:8<1664::AID-CNCR23>3.0.CO;2-G.
Chi Z, Li S, Sheng X, Si L, Cui C, Han M, et al. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: a study of 522 consecutive cases. BMC Cancer. 2011;11(1):85. https://doi.org/10.1186/1471-2407-11-85.
Hillenbrand A, Barth TF, Henne-Bruns D, Formentini A. Anorectal amelanotic melanoma. Color Dis. 2008;10(6):612–5. https://doi.org/10.1111/j.1463-1318.2007.01400.x.
Callahan A, Anderson WF, Patel S, Barnholtz-Sloan JS, Bordeaux JS, Tucker MA, et al. Epidemiology of anorectal melanoma in the United States: 1992 to 2011. Dermatol Surg. 2016;42(1):94–9. https://doi.org/10.1097/DSS.0000000000000579.
Coté TR, Sobin LH. Primary melanomas of the esophagus and anorectum: epidemiologic comparison with melanoma of the skin. Melanoma Res. 2009;19(1):58–60. https://doi.org/10.1097/CMR.0b013e32831ef262.
Tchelebi L, Guirguis A, Ashamalla H. Rectal melanoma: epidemiology, prognosis, and role of adjuvant radiation therapy. J Cancer Res Clin Oncol. 2016;142(12):2569–75. https://doi.org/10.1007/s00432-016-2245-x.
Bello DM, Smyth E, Perez D, Khan S, Temple LK, Ariyan CE, et al. Anal versus rectal melanoma: does site of origin predict outcome? Dis Colon Rectum. 2013;56(2):150–7. https://doi.org/10.1097/DCR.0b013e31827901dd.
Chen H, Cai Y, Liu Y, He J, Hu Y, Xiao Q, et al. Incidence, surgical treatment, and prognosis of anorectal melanoma from 1973 to 2011: a population-based SEER analysis. Medicine. 2016;95(7):e2770. https://doi.org/10.1097/MD.0000000000002770.
Belli F, Gallino GF, Lo Vullo S, Mariani L, Poiasina E, Leo E. Melanoma of the anorectal region: the experience of the National Cancer Institute of Milano. Eur J Surg Oncol. 2009;35(7):757–62. https://doi.org/10.1016/j.ejso.2008.05.001.
Cui C, Tang B, Guo J. Chemotherapy, biochemotherapy and anti-VEGF therapy in metastatic mucosal melanoma. Chin Clin Oncol. 2014;3(3):36. https://doi.org/10.3978/j.issn.2304-3865.2014.07.02.
David AW, Perakath B. Management of anorectal melanomas: a 10-year review. Trop Gastroenterol. 2007;28(2):76–8.
Silva IP, Long GV. Systemic therapy in advanced melanoma: integrating targeted therapy and immunotherapy into clinical practice. Curr Opin Oncol. 2017;29(6):484–92. https://doi.org/10.1097/CCO.0000000000000405.
Dumaz N, Jouenne F, Delyon J, Mourah S, Bensussan A, Lebbe C. Atypical BRAF and NRAS mutations in mucosal melanoma. Cancers (Basel). 2019;11(18):1133. https://doi.org/10.3390/cancers11081133.
Luo Y, Zhang Z, Liu J, Li L, Xu X, Yao X, et al. Characterizations of gene alterations in melanoma patients from Chinese population. Biomed Res Int. 2020. e-pub ahead of print 30 Jan 2020. https://doi.org/10.1155/2020/6096814.
Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat Commun. 2019;10(1):3163. https://doi.org/10.1038/s41467-019-11107-x.
Nassar KW, Tan AC. The mutational landscape of mucosal melanoma. Semin Cancer Biol. 2020;61:139–48. https://doi.org/10.1016/j.semcancer.2019.09.013.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. https://doi.org/10.1101/gr.107524.110.
He X, Chen S, Li R, Han X, He Z, Yuan D, et al. Comprehensive fundamental somatic variant calling and quality management strategies for human cancer genomes. Brief Bioinform. 2020;8:bbaa083.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–2. https://doi.org/10.1093/bioinformatics/btv710.
Hao M, Zhao G, Du X, Yang Y, Yang J. Clinical characteristics and prognostic indicators for metastatic melanoma: data from 446 patients in North China. Tumour Biol. 2016;37(8):10339–48. https://doi.org/10.1007/s13277-016-4914-4.
Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545(7653):175–80. https://doi.org/10.1038/nature22071.
Zou Z, Ou Q, Ren Y, Lv Q, Qin L, Zhao L, et al. Distinct genomic traits of acral and mucosal melanomas revealed by targeted mutational profiling. Pigment Cell Melanoma Res. 2020;33(4):601–11. https://doi.org/10.1111/pcmr.12865.
Carvajal RD, Spencer SA, Lydiatt W. Mucosal melanoma: a clinically and biologically unique disease entity. J Natl Compr Cancer Netw. 2012;10(3):345–56. https://doi.org/10.6004/jnccn.2012.0034.
Nagarajan P, Piao J, Ning J, Noordenbos LE, Curry JL, Torres-Cabala CA, et al. Prognostic model for patient survival in primary anorectal mucosal melanoma: stage at presentation determines relevance of histopathologic features. Mod Pathol. 2020;33(3):496–513. https://doi.org/10.1038/s41379-019-0340-7.
Cosgarea I, Ugurel S, Sucker A, Livingstone E, Zimmer L, Ziemer M, et al. Targeted next generation sequencing of mucosal melanomas identifies frequent NF1 and RAS mutations. Oncotarget. 2017;8(25):40683–92. https://doi.org/10.18632/oncotarget.16542.
Yang HM, Hsiao SJ, Schaeffer DF, Lai C, Remotti HE, Horst D, et al. Identification of recurrent mutational events in anorectal melanoma. Mod Pathol. 2017;30(2):286–96. https://doi.org/10.1038/modpathol.2016.179.
Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE, et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017;27(3):189–99. https://doi.org/10.1097/CMR.0000000000000345.
Zhou R, Shi C, Tao W, Li J, Wu J, Han Y, et al. Analysis of mucosal melanoma whole-genome landscapes reveals clinically relevant genomic aberrations. Clin Cancer Res. 2019;25(12):3548–60. https://doi.org/10.1158/1078-0432.CCR-18-3442.
Mikkelsen LH, Maag E, Andersen MK, Kruhøffer M, Larsen AC, Melchior LC, et al. The molecular profile of mucosal melanoma. Melanoma Res. 2020;30(6):533–42. https://doi.org/10.1097/CMR.0000000000000686.
Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. https://doi.org/10.1016/S0140-6736(12)60868-X.
Lee KH, Goh J, Kim YJ. Identification of synthetic chemosensitivity genes paired with BRAF for BRAF/MAPK inhibitors. Sci Rep. 2020;10(1):20001. https://doi.org/10.1038/s41598-020-76909-2.
Young K, Minchom A, Larkin J. BRIM-1, −2 and −3 trials: improved survival with vemurafenib in metastatic melanoma patients with a BRAF(V600E) mutation. Future Oncol. 2012;8(5):499–507. https://doi.org/10.2217/fon.12.43.
Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20(6):963–9. https://doi.org/10.1016/j.molcel.2005.10.022.
Quek C, Rawson RV, Ferguson PM, Shang P, Silva I, Saw RPM, et al. Recurrent hotspot SF3B1 mutations at codon 625 in vulvovaginal mucosal melanoma identified in a study of 27 Australian mucosal melanomas. Oncotarget. 2019;10(9):930–41. https://doi.org/10.18632/oncotarget.26584.
Forschner A, Hilke FJ, Bonzheim I, Gschwind A, Demidov G, Amaral T, et al. MDM2, MDM4 and EGFR amplifications and hyperprogression in metastatic acral and mucosal melanoma. Cancers (Basel). 2020;12(3):540. https://doi.org/10.3390/cancers12030540.
Hilke FJ, Sinnberg T, Gschwind A, Niessner H, Demidov G, Amaral T, et al. Distinct mutation patterns reveal melanoma subtypes and influence immunotherapy response in advanced melanoma patients. Cancers (Basel). 2020;12(9):2359. https://doi.org/10.3390/cancers12092359.
Parrella P, Caballero OL, Sidransky D, Merbs SL. Detection of c-myc amplification in uveal melanoma by fluorescent in situ hybridization. Comp Study. 2001;42(8):1679–84.
McCarthy C, Kalirai H, Lake SL, Dodson A, Damato BE, Coupland SE. Insights into genetic alterations of liver metastases from uveal melanoma. Pigment Cell Melanoma Res. 2016;29(1):60–7. https://doi.org/10.1111/pcmr.12433.
Lin X, Sun R, Zhao X, Zhu D, Zhao X, Gu Q, et al. C-myc overexpression drives melanoma metastasis by promoting vasculogenic mimicry via c-myc/snail/Bax signaling. J Mol Med. 2017;95(1):53–67. https://doi.org/10.1007/s00109-016-1452-x.
Zhang K, Wong P, Duan J, Jacobs B, Borden EC, Bedogni B. An ERBB3/ERBB2 oncogenic unit plays a key role in NRG1 signaling and melanoma cell growth and survival. Pigment Cell Melanoma Res. 2013;26(3):408–14. https://doi.org/10.1111/pcmr.12089.
Buac K, Xu M, Cronin J, Weeraratna AT, Hewitt SM, Pavan WJ. NRG1 / ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res. 2009;22(6):773–84. https://doi.org/10.1111/j.1755-148X.2009.00616.x.
Chua YL, Ito Y, Pole JC, Newman S, Chin SF, Stein RC, et al. The NRG1 gene is frequently silenced by methylation in breast cancers and is a strong candidate for the 8p tumour suppressor gene. Oncogene. 2009;28(46):4041–52. https://doi.org/10.1038/onc.2009.259.
Capparelli C, Purwin TJ, Heilman SA, Chervoneva I, McCue PA, Berger AC, et al. ErbB3 targeting enhances the effects of MEK inhibitor in wild-type BRAF/NRAS melanoma. Cancer Res. 2018;78(19):5680–93. https://doi.org/10.1158/0008-5472.CAN-18-1001.
Capparelli C, Rosenbaum S, Berger AC, Aplin AE. Fibroblast-derived neuregulin 1 promotes compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J Biol Chem. 2015;290(40):24267–77. https://doi.org/10.1074/jbc.M115.657270.
Marranci A, Jiang Z, Vitiello M, Guzzolino E, Comelli L, Sarti S, et al. The landscape of BRAF transcript and protein variants in human cancer. Mol Cancer. 2017;16(1):85. https://doi.org/10.1186/s12943-017-0645-4.
Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3(1):724. https://doi.org/10.1038/ncomms1727.
Lezcano C, Shoushtari AN, Ariyan C, Hollmann TJ, Busam KJ. Primary and metastatic melanoma with NTRK fusions. Am J Surg Pathol. 2018;42(8):1052–8. https://doi.org/10.1097/PAS.0000000000001070.
Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–9. https://doi.org/10.1158/2159-8290.CD-16-1237.
Kim HS, Jung M, Kang HN, Kim H, Park CW, Kim SM, et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene. 2017;36(23):3334–45. https://doi.org/10.1038/onc.2016.486.
Acknowledgments
Not applicable.
Funding
This study was supported by the Beijing Municipal Science and Technology Commission NOVA program (NO.2010 B033), the Beijing Municipal Science and Technology Commission Capital Characteristic Clinical Application Research (No. Z141107002514077), Capital’s Funds for Health Improvement and Research 2018–2-1022 and the National Nature Science Foundation of China (No.81773214).
Author information
Authors and Affiliations
Contributions
Z.L. and L.S. conceived and designed the experiments. H. L, L. Y, Y.L., D.W., X. L, N. H and Y. K performed the experiments. H.L., L.Y. and X.W. analyzed the data. D.W., X. L and X. H contributed the materials/analysis tools for the study. H.L. and Y.L. updated the follow-up data for all cases. H.L., X.W. and X.H. wrote the manuscript. H.L., X.H., S.L. and X.H. prepared figures and tables. All authors have read and approved the final manuscript. Co-published data notes will be linked to the research article the data support.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All analyses of human data conducted in this study were approved by the Ethics Committee of Peking University Cancer Hospital and all methods were carried out in accordance with relevant guidelines and regulations. Written informed consent was obtained from all participants. This study was performed in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
None of the authors have any competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Somatic mutations in 36 RMM.
Additional file 2: Table S2.
CNVs and SVs variants in 36 RMM.
Additional file 3: Table S3.
Univariate analysis to identify OS-related clinicopathological features.
Additional file 4: Figure S1.
Distribution of RMM somatic mutations in the protein. Somatic mutation sites identified in our study were shown in functional domains, the amino acid length of Lollipop graph with indicates the number of mutations.
Additional file 5: Figure S2.
The overview of identifying OS-related factors in 36 RMM patients. RMM patients were analyzed through the TSO500 pipeline and 388 mutations were identified. Univariate and multivariate regression analysis were performed to identify prognostic factors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, H., Yang, L., Lai, Y. et al. Genetic alteration of Chinese patients with rectal mucosal melanoma. BMC Cancer 21, 623 (2021). https://doi.org/10.1186/s12885-021-08383-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08383-6