
Abstract
Background
Triple-negative breast cancer (TNBC) is a heterogeneous disease and we have previously shown that rapid relapse of TNBC is associated with distinct sociodemographic features. We hypothesized that rapid versus late relapse in TNBC is also defined by distinct clinical and genomic features of primary tumors.
Methods
Using three publicly-available datasets, we identified 453 patients diagnosed with primary TNBC with adequate follow-up to be characterized as ‘rapid relapse’ (rrTNBC; distant relapse or death ≤2 years of diagnosis), ‘late relapse’ (lrTNBC; > 2 years) or ‘no relapse’ (nrTNBC: > 5 years no relapse/death). We explored basic clinical and primary tumor multi-omic data, including whole transcriptome (n = 453), and whole genome copy number and mutation data for 171 cancer-related genes (n = 317). Association of rapid relapse with clinical and genomic features were assessed using Pearson chi-squared tests, t-tests, ANOVA, and Fisher exact tests. We evaluated logistic regression models of clinical features with subtype versus two models that integrated significant genomic features.
Results
Relative to nrTNBC, both rrTNBC and lrTNBC had significantly lower immune signatures and immune signatures were highly correlated to anti-tumor CD8 T-cell, M1 macrophage, and gamma-delta T-cell CIBERSORT inferred immune subsets. Intriguingly, lrTNBCs were enriched for luminal signatures. There was no difference in tumor mutation burden or percent genome altered across groups. Logistic regression mModels that incorporate genomic features significantly outperformed standard clinical/subtype models in training (n = 63 patients), testing (n = 63) and independent validation (n = 34) cohorts, although performance of all models were overall modest.
Conclusions
We identify clinical and genomic features associated with rapid relapse TNBC for further study of this aggressive TNBC subset.
Similar content being viewed by others

Background
Triple negative breast cancer (TNBC) is an aggressive breast cancer subtype defined by lack of targetable estrogen receptor (ER), progesterone receptor (PR), and HER2 [1]. TNBC accounts for 15% of breast cancer cases, yet is responsible for 35% of breast cancer related deaths [1, 2]. Relative to hormone receptor positive breast cancers, TNBCs are more likely to develop distant rather than local recurrence and TNBCs spread more frequently to visceral sites, including lung and brain [2,3,4]. Understanding determinants of distant relapse is imperative as the median overall survival after diagnosis of metastatic disease was historically only 13–17 months [2, 5] and remains only 25 months even among patients with PD-L1 positive TNBC receiving chemo-immunotherapy [6].
Advances in sequencing technology have facilitated comprehensive molecular profiling of breast cancers, including subsets of TNBC [7, 8]. Two landmark analysis of primary TNBCs revealed six subtypes of TNBC with distinct expression profiles [9, 10] and an integrated copy number/transcriptome analysis identified four overlapping TNBC subsets [11]. Genomic analyses demonstrate high frequency of mutations in TP53 (~ 75% of TNBCs) and PIK3CA ~ 25% [11,12,13] while TNBCs also reflect widespread copy number alterations [11,12,13]. The existing TNBC subsets/groupings provide a critical framework for understanding intrinsic genomic characteristics but are only associated with modest differences in patient survival. Among the approximately 30% of TNBCs who develop metastatic disease, a subset have an aggressive phenotype associated with rapid relapse, therapeutic resistance, and poor prognosis, while others have a relatively late relapse associated with more indolent or treatment responsive disease – yet we have a poor understanding of genomic features associated with distinct timing of relapse [1, 2, 14].
To more accurately understand the differences in patient outcome in TNBC, we sought to understand distinct clinical and genomic features among primary TNBCs categorized based on outcome: rapid (rrTNBC), late (lrTNBC) and no relapse (nrTNBC). In several large TNBC cohort studies, the median time to distant metastasis was around 2 years, ranging from 19.7 to 31.2 months, [2, 14,15,16] thus we define rrTNBC as relapse or death within 24 months of diagnosis. We previously demonstrated in two large cohorts (Surveillance, Epidemiology, and End Results Program/SEER and National Comprehehensive Cancer Network/NCCN) that disparities in sociodemographic features are strongly associated with rrTNBC, including insurance type, race, and surgical management [17,18,19]. These studies demonstrate the relevance of understanding factors contributing to rrTNBC yet are limited by lack of biologic understanding.
As an initial investigation of genomic features associated with rrTNBC, we aggregated data across multiple cohorts then utilized a train/test split and an independent validation cohort to model predictors of rapid versus late relapse.
Methods
Patient and tumor characteristics
Patient-specific data were obtained from The Cancer Genome Atlas (TCGA) [12], Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) [20, 21], our published meta-analysis (“neoadjuvant dataset” as described previously) [7], and the Fudan TNBC cohort [22]. These variables included age at diagnosis, grade, stage at diagnosis, pathologic receptor status (ER, PR, and HER2), response to neoadjuvant chemotherapy (when available), and distant metastasis-free or overall survival. TNBC was defined as being negative for ER, PR, and HER2: immunohistochemistry (IHC) 0 and FISH HER2/CEP17 ratio of less than 2.0. Neoadjuvant chemotherapy response was based on study-reported outcomes. As we previously reported, all patients in the “neoadjuvant dataset” received neoadjuvant chemotherapy but from diverse regimens: 41% of patients received anthracycline/taxane +/− alkylator, 15% anthracycline +/− alkylator, 35% taxane alone, and 9% anthracycline/platinum.
Genomic data
For data from the METABRIC, normalized gene expression data, copy number data, and somatic mutation data for 171 cancer-related genes were obtained from the publicly available European Genome-Phenome Archive (IDs EGAD00010000210 and EGAD0001000021) and associated publications [13, 21]. Copy number segmented data files were processed using GISTIC2.0 [23]. For data from TCGA, breast cancer gene expression data, GISTIC copy number data, and somatic mutation data were obtained from the XENAbrowser (version 2015-02-24). Gene expression data from 17 published studies of breast cancer patients prior to NAC were re-processed from raw files, as previously described [7]. Genomic data from the Fudan TNBC study was downloaded from the National Omics Data Encyclopedia (accession OEP000155) [22].
Gene expression signatures, expression-based subtypes, and inferred immune subsets
Given gene expression data from multiple studies and disparate platforms, gene expression data for all TNBCs for each dataset (METABRIC n = 287, TCGA n = 160, neoadjuvant dataset n = 446) were extracted, quantile normalized within TNBCs from each study, and subsequently median centered. We evaluated summary expression metrics (e.g. signatures, intrinsic subtypes, CIBERSORT proportions). One hundred twenty-five published gene expression signatures were calculated as we have previously described [7]. We determined PAM50 intrinsic breast cancer subtype using the ‘Bioclassifier’ package from Parker et al. after balancing TNBC data with an equal number of ER-positive cases for each dataset [24]. TNBC subtype was determined using the TNBCtype tool [9, 25]. Proportion of infiltrating immune cell subsets were calculated using the CIBERSORT algorithm [26].
Modeling and performance
We compared the performance of three logistic regression models in predicting rapid relapse versus late relapse. The “null model” contained only clinical variables (age/stage at diagnosis and PAM50/TNBC subtype). The “null plus significant genomic features”, adds any feature significantly different between rrTNBC and lrTNBC with a nominal p-value < 0.05. The “genomic features reduced”, is a reduced version of the second model that only includes features among the top 25 most important genomic features in at least half of the independent runs. Lasso reduction and tuning of the regularization parameter lambda were performed. To evaluate model performance, we calculated the average receiver-operator characteristic (ROC) AUC of the 25 runs, and 95% confidence interval was calculated using the standard deviation of the sample of means.
Statistical analysis
Differences in patient and tumor characteristics were evaluated using Pearson chi-squared tests. The association of gene signatures with neoadjuvant chemotherapy response was evaluated using simple linear regression and t-tests. All calculations of association were multiple-testing corrected using the Benjamini–Hochberg procedure for false discovery rate. For continuous variables, we calculated p-values comparing rapid vs. late and relapse vs. no relapse using ANOVA and logistic regression. For count variables (e.g. mutated vs. not) we used Fisher exact tests to evaluate relapse vs. not and rapid vs. late relapse. P-values for CIBERSORT and mutation signatures were evaluated using logistic regression, while CNAs, and mutations were evaluated using Fisher exact tests. Data visualization was made using ggplot2 [27]. All statistical analyses were performed in R version 3.4.1.
Results
Defining rapid vs. late vs. no relapse triple-negative breast cancer
From three large cohorts with primary breast cancer genomic data – TCGA, [12] METABRIC, [20, 21] and our prior breast cancer gene expression meta-analysis [7] – we identified 893 TNBCs from a total of 4473 breast cancer cases. For our analyses, we included patients with at least 60 months of follow-up or those with a distant metastasis-free survival (DMFS) event prior to our 60-month cutoff, leaving a total of 453 TNBCs in our evaluable dataset. Of these, 453 had gene expression data, 317 had copy number data, and 317 had mutation data. (Fig. 1a).

Study design and definition of triple-negative breast cancer (TNBC) rapid vs. late relapse. a REMARK diagram. b Proportion of distant metastasis-free survival (DMFS) events per year after diagnosis among evaluable dataset. ‘Rapid relapse’ was defined as DMFS events within the 2 years of diagnosis and ‘late relapse’ DMFS events beyond 2 years. c-e Kaplan-Meier diagram of DMFS in study cohort reflecting TNBC group definitions (c), compared with DMFS by intrinsic subtype approaches PAM50 subtype (d), and Lehmann TNBC subtype (e). P-value indicates log-rank test
We assessed the percentage of total DMFS events each year (Fig. 1b). In this dataset, over 20% of DMFS events occurred each of the first 2 years after diagnosis, categorized as ‘rapid relapse’ (rrTNBC). Among lrTNBCs, most DMFS events occurred within the first 5 years after diagnosis, with sporadic events beyond year 6. Our main goal was to identify differences among TNBCs with clinically distinct outcomes, so we visualized DMFS for our relapse categorization (Fig. 1c) in comparison with DMFS for existing intrinsic expression-based subtype approaches PAM50 [24] (Fig. 1d) or Lehmann/Pietenpol TNBCtype [9] (Fig. 1e) within the same cohort. The Lehmann/Pietenpol TNBCtype (log-rank p = 0.01), but not PAM50, was associated with significant differences in DMFS. The strikingly different visualized outcomes suggests that our relapse categorization does, in fact, identify truly distinct subsets based on outcome when compared to approaches that focus on intrinsic features.
Patient and tumor characteristics
We evaluated the association of clinical, pathologic, and intrinsic expression subtype with rapid vs. late vs. no relapse status (Table 1). There was no significant difference in age at diagnosis or grade, however, rrTNBCs were significantly more likely to be higher stage (Chi-square p = 1.9e-10). The majority of patients were basal-like PAM50 subtype (78%), but, lrTNBCs were significantly more likely to be non-basal (non-basal: rrTNBC 18%, lrTNBC 29%, nrTNBC 20%, Chi-square p = 0.03). Lehmann/Pietenpol TNBC subtype also reflected significant differences across groups (Chi-square p = 0.02). The immunomodulatory phenotype was highest in nrTNBC (16% rrTNBC, 16% lrTNBC, 24% nrTNBC), luminal androgen receptor was highest in lrTNBC (9% rrTNBC, 16% lrTNBC, 9% nrTNBC), and basal-like 2 was highest in rrTNBC (15% rrTNBC, 9% lrTNBC, 6% nrTNBC). A subset of patients in this cohort (127/453; 28.0%) had data on response to neoadjuvant chemotherapy (NAC). As anticipated, those patients with rrTNBC or lrTNBC were significantly more likely to have residual disease (RD) after neoadjuvuant chemotherapy (93 and 94% RD, respectively), relative to those with nrTNBC (51% RD; Chi-square p = 1.9e-7). Intriguingly, the rate of residual disease was similar among rrTNBC and lrTNBC despite markedly different timing of relapse.
Response to Neoadjuvant chemotherapy and survival in TNBC: immune and expression signatures
Response to NAC is known to be a robust prognostic biomarker in TNBC [28]. In this cohort, only 28% (127/453) of patients received NAC and many of the regimens were non-standard (e.g. taxane alone). Because of this, the pathologic complete response (pCR) after NAC was only 22.8%, much lower than modern current regimens, typically ~ 40%. Despite these significant limitations, pCR was strongly associated with nrTNBC (p < 0.001). The patients with data on response to NAC all had whole transcriptome data but no available mutation or copy number data, so we calculated a score for 125 published gene expression signatures and evaluated the association of each signature with NAC response (pCR vs. RD) and DMFS. Signatures were grouped by phenotype as previously described [7] (n = 127 patients; Fig. 2a). Immune signatures were associated with better prognosis and most were also associated with improved response to NAC. Proliferation signatures tended to be associated with improved response to NAC, as we have previously described [7], yet there was variable association with DMFS.

Immune and Expression Signatures and Response to Neoadjuvant Chemotherapy and Survival in TNBC. a The calculated score for 125 published gene expression signatures for 127 patients with data on response to neoadjuvant chemothrapy and distant metastasis-free survival (DMFS). Each signature is a point. The association of each signature with neoadjuvant chemotherapy response (pathologic complete response vs. RD) by simple linear regression (y-axis) and hazard ratio for each signature using DMFS (x-axis) are displayed. Signatures were grouped by phenotype (as previously described [7]), identified by color: proliferation signatures (red), immune signatures (blue), ER/HER2 signatures (green), mesenchymal signatures (orange), others (grey). Size of each point relates to the hazard ratio p-value for each signature. b The association of three representative signatures from each group (immune, proliferation, ER/HER2, mesenchymal) with the relative proportion of 22 inferred immune cell subsets via CIBERSORT across all samples with gene expression data (n = 453) are visualized using CorrPlot [26, 29].
To understand what immune cell types in the tumor microenvironment may be reflected by the immune signatures, we visualized the association of three representative signatures from each group (immune, proliferation, ER/HER2, mesenchymal) with the relative proportion of 22 inferred immune cell subsets via CIBERSORT (Fig. 2b) [26]. Immune signatures were strongly positively correlated with anti-tumor immune cell types including M1 macrophages, CD8 T-cells, and memory B-cells (all Pearson’s r ≥ 0.3, all p < 1.2e-8) and anti-correlated with immune suppressive cell types including M2 macrophages, memory resting CD4 T-cells, resting NK cells, and resting mast cells. ER/HER2 signatures reflected an almost opposite pattern to immune signatures, with positive correlation to immune suppressive cell types and anti-correlation with anti-tumor immune cell type. Metabolic signatures appeared to have a strong correlation specifically with M0 macrophages (all Pearson’s r > 0.27, all p < 8.4e-9). As a sensitivity analysis, we evaluated the association of three representative signatures from each group with 7 immune cell-type specific signatures from MSigDB [30, 31] (instead of CIBERSORT) and found similar results (Supplementary Figure 1A).
Expression signatures in rapid vs. late vs. no relapse TNBC
To assess pathways and phenotypes associated with rapid vs. late vs. no relapse, a score was calculated for 125 published gene expression signatures across the entire dataset (Supplementary Figure 1B). Evaluating each signature individually across the three groups revealed 16 signatures that were significantly different (ANOVA FDR p < 0.05; Fig. 3, Supplementary Figure 2A-B). Among these, five signatures were immune-related [9, 32,33,34] and all were significantly higher in nrTNBC relative to rrTNBC and lrTNBC. Eight significant signatures were related to luminal phenotype – all were highest in lrTNBC, lowest in rrTNBC, and intermediate in nrTNBC. While we and others have demonstrated that proliferation signatures are strongly associated with response to neoadjuvant chemotherapy independent of immunophenotype [7, 35] as well as overall survival, [36] we did not identify a significant association of proliferation signatures across all three groups (Supplementary Figure 2B). However, when evaluating rapid versus late relapse only as an exploratory analysis, late relapse was associated with significantly lower proliferation, for example the PAM50 proliferation score (t-test p = 0.007). Most CIBERSORT immune subsets were not statistically significant (Supplementary Figure 2C), however, neutrophils were significantly higher in rrTNBC (ANOVA FDR p = 0.001). To more comprehensively investigate inferred immune subsets, we evaluated the association of summed protumorigenic subsets (Tcells-CD4 naive, Bcells-naive, Mast cells-resting, NK cells-resting, Tcells-CD4 memory resting, Plasma cells, Dendritic cells resting, Tcells-regulatory/Tregs, Macrophages-M0, MacrophagesM2) and summed antitumorigenic subsets (Monocytes, Eosinophils, Tcells-gamma delta, Tcells-follicular helper, Tcells-CD8, NK cells-activated, Bcells-memory, Mast cells-activated, Neutrophils, Macrophages-M1, Dendritic cells-activated, Tcells-CD4 memory activated) with rapid versus late versus no relapse (Supplementary Figure 2D). Antitumorigenic subsets were significantly different among relapse groups (ANOVA p = 0.002), highest in ‘no relapse’, while there was no difference in the protumorigenic subsets (ANOVA p = 0.62), although the absolute differences were small.

Expression Signatures in Rapid vs. Late vs. No Relapse TNBC. The calculated score for 16 published gene expression signatures that demonstrated statistical significance (ANOVA FDR p < 0.05) comparing rapid vs. late vs. no relapse. Signatures visualized as relative values (Z-score) with rapid relapse (red), late relapse (green), and no relapse (blue)
Mutations and copy number alterations
In this cohort, 70% (317/453) of patients had data on single nucleotide variant/mutation data including 171 cancer-related genes and whole genome CNAs [21]. Only a small subset of patients (11.7%; 53/453) had whole exome mutation data, so we focused on the 171 cancer-related genes to ensure adequate statistical power. When evaluating general mutational features, there was no significant difference in mutations per megabase (ANOVA p = 0.64; Fig. 4a) nor percent genome altered by copy number (ANOVA p = 0.96; Fig. 4b).

Mutations and copy number alterations in rapid vs. late vs. no relapse TNBCs. a Mutations per megabase of 171 cancer-related genes. b Percent genes altered by copy number gain (GISTIC 1 or 2) or loss (GISTIC −1 or − 2). c Frequency of alteration of 171 cancer-related genes (green dots), copy number gains (red dots) or losses (red dots) by cytoband among rapid relapse (x-axis) vs. no relapse (y-axis) TNBCs (c) or rapid relapse (x-axis) vs. late relapse (y-axis) TNBCS (d). Size of dot indicates negative log of p-value for Fisher exact test with those genes and cytobands indicated demonstrate nominal p < 0.05. Zoomed-in image of those alterations with < 20% frequency indicated in right panel
We first compared the frequency of alteration for each mutation and cytoband (for CNAs) for relapse (rrTNBC + lrTNBC) vs. nrTNBC (Fig. 4c) because of low mutation frequency for most genes. There were no genes that were significantly different after multiple testing (Supplementary Figure 3A) when comparing relapse vs. no relapse, but PIK3CA mutations were more frequent in relapse relative to nrTNBC. In addition, PTEN, ARID1A, and RYR2 mutations were enriched in nrTNBC relative to rrTNBC (Fisher exact nominal p < 0.05). We then compared rrTNBC vs. lrTNBC (Fig. 4d) and found that rrTNBC were significantly more likely to harbor a mutation in TP53 compared to lrTNBC patients (Fisher exact FDR p = 0.009). Among CNAs, the copy number landscape was similar across the rapid vs. late vs. no relapse groups (Supplementary Figure 3B) and there were no significantly altered genes or regions among these three groups after multiple test correction yet there were several regions that demonstrated enrichment within specific groups (nominal p < 0.05; Fig. 4c-d).
Clinical and multi-‘omic model of rapid vs. late relapse in TNBC
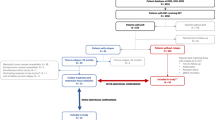
Having identified discrete clinical, expression, immune, mutation, and copy number features among primary TNBCs with distinct clinical outcomes, we sought to develop an optimal, multi-‘omic predictive model for rrTNBC vs. lrTNBC. We compared performance of three logistic regression models with lasso reduction (detailed in the Methods; Fig. 5a). The clinical, “null model”, performed marginally in both the testing cohort and the independent validation cohort (average AUC 0.574 and 0.525, respectively). The other two models (clinical+genomic and reduced genomic) had significantly improved performance in both the testing cohort and the independent validation (average AUC: 0.774 and 0.821 for testing; 0.645 and 0.620 for validation; Fig. 5b; all Wilcoxon rank sum p < 0.005). The genomic features that contributed most included clinical features (stage, expression subtypes), mutations (ARID2, DNAH11, SETDB1), copy number alterations (loss LAMA2, CLK3, MLLT4, SYNE1 and gain DNAH5, LIFR, PETN), and expression signatures (signatures of RBBP8 [37], ER negative chemoresistance [38], PTEN deletion [39], beta catenin [40], STAT3 [41], and RAS pathway activation [42]). We evaluated additional models, including machine learning approaches (random forest, support vector machine) as well as the universe of available genomic data, however, these additional modeling approaches were characterized by overfitting even in the context of model tuning and demonstrated no significant improvement in performance relative (data not shown).

Developing an optimal clinical and multi-‘omic model of rapid vs. late relapse in TNBC. a Schematic of experimental steps including definition of variables, descriptive statistics, comparative modeling including model tuning, and assessment of model performance. b Receiver-operator characteristic (ROC) plots for each model’s performance, measured by average area under the curve (AUC) of 25 independent runs of the train-test split. Each model was tuned to ensure optimal performance. Models are grouped and colored by cohort—red indicates training data (n = 63), green indicates testing data (n = 63), and blue indicates the independent validation Fudan cohort (n = 34). For each grouping, the three models shown are: 1) “null model”, including only clinical variables; 2) “null plus significant features”, adding any feature significantly different between rrTNBC and lrTNBC with a nominal p-value < 0.05; and 3) “null plus significant features reduced”, including only features from model 2 that are among the top 25 most important genes in at least half of the independent runs. Asterisks indicate significance by Wilcoxon rank sum, * indicates p < 0.05, ** indicates p < 0.01, NS indicates “not significant” (p > 0.05)
Discussion
We previously demonstrated in two large cohorts that disparities in sociodemographic features are strongly associated with rrTNBC, [17,18,19] and in this report sought to investigate genomic features associated with rrTNBC. We aggregated data from the available cohorts that have multi-‘omic data as well as adequate follow-up to characterize TNBCs as rrTNBC, lrTNBC, or nrTNBC. Although limited by the retrospective nature and limited number of patients who received modern NAC, we provide initial observations regarding genomic features and rrTNBC.
Our goal was to identify distinguishing features and determine if predictive models incorporating clinical, expression-based subtype, and/or multi-‘omic models could identify patients at high risk of rapid relapse. lrTNBCs are more likely to be non-basal (primarily luminal A/B) and our data identify eight luminal signatures are associated with late relapse. Multiple groups have identified a ‘luminal androgen receptor’ subset of TNBC based on molecular classifications, [9, 11] and 40% (20/49) of the Lehmann LAR subtype tumors in our cohort ultimately had late relapse. To develop predictive models, we first identified the relatively few specific features that were significantly different across subsets (61 features from > 35,000 initial data points) then built models based on a priori feature identification. This approach led to overall good performance of multiple models, and importantly allows us to understand what genomic features contribute most. More complex modeling approaches (e.g. machine learning algorithms) did not improve model performance and led to challenges with overfitting. Our models were evaluated in over two times the number of TNBC patients available in TCGA [12] alone – a remarkable number for a disease that accounts for only approximately 15% of breast cancers [1, 2]. Collectively, our data support the categorization by Burstein et al. [11] and suggest that lrTNBCs are enriched for luminal phenotypes while rrTNBCs are likely enriched for the ‘basal-like immune suppressed’ phenotype.
Stage at diagnosis was strongly associated with rrTNBC in univariate analyses and in logistic regression models. One hypothesis is that stage at diagnosis captures non-biological features including socioeconomic or demographics features [43,44,45]. Race/ethnicity is complex, [46, 47] was largely unavailable in the included datasets, and warrants further study [48, 49]. In a parallel study, we investigated the association of sociodemographic features with rrTNBC among 3016 primary TNBCs at ten academic cancer centers [50]. In this large cohort, we found that stage at diagnosis remained significant, as well as Medicaid/indigent insurance, lower income, and younger age [50]. Collectively, this suggests that timing of relapse is impacted by a complex set of clinical, genomic, and sociodemographic features that warrant further multi-level analyses.
Response to neoadjuvant chemotherapy remains the best prognostic biomarker for TNBC, [28] but there are clear differences in disease course among TNBCs who develop relapse earlier vs. later. At the time of these analyses, no large multi-‘omic dataset including NAC and long-term outcomes were available although this is anticipated in the future. Despite significant limitations of NAC analyses, somewhat unexpectedly patients destined for rrTNBC and lrTNBC in this cohort had similarly high rates of residual disease to neoadjuvant chemotherapy. Both rrTNBC and lrTNBC had lower expression of immune signatures compared with nrTNBCs, reflecting reduced anti-tumor immune response. This supports our and others’ work, [7, 51,52,53,54] including our analyses of the BrighTNess phase III clinical trial, which provides largest transcriptome dataset and association with NAC and demonstrated that stratifying patients by proliferation and immune signatures can effectively stratify likelihood of pCR irrespective of NAC regimen. Given the recent FDA approval of immunotherapy for metastatic TNBC [6], there is great interest to augment the existing host anti-tumor immune response [55,56,57,58].
Clinically, it is clear that a subset of patients with TNBC have highly aggressive, largely treatment-refractory disease [1, 2, 14]. In the modern era, NAC offers a biological ‘readout’ of chemosensitivity that is highly associated with both recurrence and survival endpoints and has become standard of care, with pathologic response used to guide subsequent escalation/de-escalation of adjuvant therapy [59]. However, among the highest risk TNBCs with RD after NAC, we still have limited ability to identify the ~ 40% patients destined for relapse [7, 8]. We envision that the results of this and similar efforts, such as circulating tumor DNA minimal residual disease assays, [60,61,62] could identify patients at highest risk (rrTNBCs in the current study) and direct these patients to escalation of therapy, additional maintenance therapy, and/or intensive monitoring.
While this study presents promising methods to categorize TNBC relapse it does possess significant limitations. Categorization of tumors depends on study-reported estrogen receptor (ER) status; variability and changes in standard determination of estrogen receptor positivity since 2010 guidelines [63] may have influenced whether a subset of tumors included had very low ER (e.g. < 10%). The lack of available robust multi-‘omic datasets with long-term outcome data leads to inherent limitations of aggregating multiple datasets. We incorporated genomic data from multiple studies, generated using multiple platforms, and over multiple years. While we have attempted to account for this through standard normalization approaches and analysis only of summary statistics (e.g. expression signatures not individual genes), batch/platform effects and computational analyses could impact our results. For assessment of tumor mutation burden, we used mutation data from a 317 gene targeted panel assay. While several studies suggest that TMB by targeted panel overall correlates with whole exome or whole genome sequencing, these methodologies are not identical [64,65,66]. Therapy for TNBC has changed, including: 1) standard use of neoadjuvant chemotherapy for nearly all patients with TNBC, while not all patients included received neoadjuvant or adjuvant therapy, particularly in METABRIC (161/273; 59.0%); 2) incorporation of capecitabine for RD based on CREATE-X [67]; and 3) recent FDA approval of immunotherapy for metastatic, PD-L1 positive TNBC [6].
In conclusion, we provide evidence that rrTNBC reflects a distinct clinical entity characterized by unique genomic features. Predictive modeling using clinical and genomic features in these datasets revealed modest results, but with improved data may identify patients at high risk for ‘rapid relapse.’ Multi-level analyses of the interaction between clinical, multi-‘omic, and sociodemographic features and timing of relapse are warranted.
Availability of data and materials
Data that support the findings of this study have been deposited in the following repositories.
Neoadjuvant dataset: Raw gene expression data and paired clinical feature data were obtained from NCBI Gene Expression Omnibus (GEO) via accession numbers GSE8465, GSE16446, GSE18728, GSE19697, GSE20194, GSE20271, GSE21974, GSE21997, GSE22093, GSE22226, GSE22358, GSE22513, GSE23988, GSE25066, GSE28796, and GSE32646).
METABRIC: Molecular Taxonomy of Breast Cancer International Consortium. Normalized gene expression data, copy number data, and paired clinical feature data were obtained from the publicly available European Genome-phenome Archive (IDs EGAD00010000210 and EGAD0001000021) with mutation data from Pereira, et al. [21].
TCGA: Normalized gene expression data, copy number data, single nucleotide variant, and paired clinical feature data TCGA data (2015-02-24 datafreeze) were obtained from the University of California, Santa Cruz cancer browser (https://xenabrowser.net/heatmap/).
Fudan dataset: Data can be viewed in The National Omics Data Encyclopedia (NODE) (http://www.biosino.org/node), accession OEP000155. Microarray data and sequence data are available in NCBI GO (OncoScan array; GEO: GSE118527) and Sequence Read Archive (WES and RNA-seq; SRA: SRP157974).
Abbreviations
- TNBC:
-
Triple-negative breast cancer
- rrTNBC:
-
Relapse/death ≤2 years of diagnosis
- lrTNBC:
-
Relapse/death > 2 years
- nrTNBC:
-
>5 years no relapse/death.
- METABRIC:
-
Molecular Taxonomy of Breast Cancer International Consortium
- TCGA:
-
The Cancer Genome Atlas
- ER:
-
Estrogen receptor
- PR:
-
Progesterone receptor
- IHC:
-
Immunohistochemistry
- ROC:
-
Receiver-operator characteristic
References
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer registry. Cancer. 2007;109(9):1721–8. https://doi.org/10.1002/cncr.22618.
Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–45. https://doi.org/10.1002/cncr.23930.
Lin NU, Vanderplas A, Hughes ME, Theriault RL, Edge SB, Wong YN, et al. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the national comprehensive Cancer network. Cancer. 2012;118(22):5463–72. https://doi.org/10.1002/cncr.27581.
Haffty BG, Yang Q, Reiss M, Kearney T, Higgins SA, Weidhaas J, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24(36):5652–7. https://doi.org/10.1200/JCO.2006.06.5664.
Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, et al. Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer. 2009;9(1):29–33. https://doi.org/10.3816/CBC.2009.n.005.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast Cancer. N Engl J Med. 2018;379(22):2108–21. https://doi.org/10.1056/NEJMoa1809615.
Stover DG, Coloff JL, Barry WT, Brugge JS, Winer EP, Selfors LM. The role of proliferation in determining response to Neoadjuvant chemotherapy in breast Cancer: a gene expression-based meta-analysis. Clin Cancer Res. 2016;22(24):6039–50. https://doi.org/10.1158/1078-0432.CCR-16-0471.
Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–45. https://doi.org/10.1158/2159-8290.CD-13-0286.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. https://doi.org/10.1172/JCI45014.
Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of triple-negative breast Cancer molecular subtypes: implications for Neoadjuvant chemotherapy selection. PLoS One. 2016;11(6):e0157368. https://doi.org/10.1371/journal.pone.0157368.
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–98. https://doi.org/10.1158/1078-0432.CCR-14-0432.
The Cancer Genome Atlas. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. https://doi.org/10.1038/nature11412.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. https://doi.org/10.1038/nature10983.
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–34. https://doi.org/10.1158/1078-0432.CCR-06-3045.
van Roozendaal LM, Smit LHM, Duijsens G, de Vries B, Siesling S, Lobbes MBI, et al. Risk of regional recurrence in triple-negative breast cancer patients: a Dutch cohort study. Breast Cancer Res Treat. 2016;156(3):465–72. https://doi.org/10.1007/s10549-016-3757-4.
Ghosh J, Agarwal S, Ganguly S, Dabkara D, Biswas B, Nandi S, et al. Patterns of recurrence in triple negative breast cancer patients (automated IHC): An Indian Tertiary Care Center data. J Clin Oncol. 2018;36(15_suppl):e13128.
Obeng-Gyasi S, Asad S, Fisher JL, Rahurkar S, Stover DG. Socioeconomic and surgical disparities are associated with rapid relapse in patients with triple-negative breast cancer. Ann Surg Oncol. 2021. https://doi.org/10.1245/s10434-021-09688-3.
Obeng-Gyasi S, Stover DG. ASO author reflections: disparities in oncologic surgery: the problem we all live with. Ann Surg Oncol. 2021. https://doi.org/10.1245/s10434-021-09694-5.
Asad S, Barcenas CH, Bleicher RJ, Cohen AL, Javid SH, Levine EG, et al. Sociodemographic factors associated with rapid relapse in triple-negative breast Cancer: a multi-institution study. J Natl Compr Cancer Netw. 2021:1–8. https://doi.org/10.6004/jnccn.2020.7659.
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–9. https://doi.org/10.1038/nature10933.
Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7(1):11479. https://doi.org/10.1038/ncomms11479.
Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and Transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. 2019;35(3):428–40 e425. https://doi.org/10.1016/j.ccell.2019.02.001.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. https://doi.org/10.1200/JCO.2008.18.1370.
Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–82. https://doi.org/10.1038/bjc.2012.451.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7. https://doi.org/10.1038/nmeth.3337.
Wickam H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2009. https://doi.org/10.1007/978-0-387-98141-3.
Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–72. https://doi.org/10.1016/S0140-6736(13)62422-8.
Cardoso F, Costa A, Senkus E, Aapro M, Andre F, Barrios CH, et al. 3rd ESO-ESMO international consensus guidelines for advanced breast Cancer (ABC 3). Breast. 2017;31:244–59. https://doi.org/10.1016/j.breast.2016.10.001.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. https://doi.org/10.1073/pnas.0506580102.
Abbas AR, Baldwin D, Ma Y, Ouyang W, Gurney A, Martin F, et al. Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 2005;6(4):319–31. https://doi.org/10.1038/sj.gene.6364173.
Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33(9):983–91. https://doi.org/10.1200/JCO.2014.58.1967.
Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14(16):5158–65. https://doi.org/10.1158/1078-0432.CCR-07-4756.
Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, et al. T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res. 2009;11(2):R15. https://doi.org/10.1186/bcr2234.
Metzger Filho O, Stover DG, Asad S, Ansell PJ, Watson M, Loibl S, et al. Immunophenotype and proliferation to predict for response to neoadjuvant chemotherapy in TNBC: results from BrighTNess phase III study. Proc Am Soc Clin Oncol. 2019;37(15_suppl):510. https://doi.org/10.1200/JCO.2019.37.15_suppl.510.
Prat A, Lluch A, Albanell J, Barry W, Fan C, Chacon J, et al. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br J Cancer. 2014;111(8):1532–41. https://doi.org/10.1038/bjc.2014.444.
Furuta S, Wang JM, Wei S, Jeng YM, Jiang X, Gu B, et al. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell. 2006;10(1):13–24. https://doi.org/10.1016/j.ccr.2006.05.022.
Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA. 2011;305(18):1873–81. https://doi.org/10.1001/jama.2011.593.
Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104(18):7564–9. https://doi.org/10.1073/pnas.0702507104.
Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–7. https://doi.org/10.1038/nature04296.
Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65(12):5054–62. https://doi.org/10.1158/0008-5472.CAN-04-4281.
Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genet. 2010;3(1):26. https://doi.org/10.1186/1755-8794-3-26.
Harper S, Lynch J, Meersman SC, Breen N, Davis WW, Reichman MC. Trends in area-socioeconomic and race-ethnic disparities in breast cancer incidence, stage at diagnosis, screening, mortality, and survival among women ages 50 years and over (1987-2005). Cancer Epidemiol Biomark Prev. 2009;18(1):121–31. https://doi.org/10.1158/1055-9965.EPI-08-0679.
Booth CM, Li G, Zhang-Salomons J, Mackillop WJ. The impact of socioeconomic status on stage of cancer at diagnosis and survival: a population-based study in Ontario, Canada. Cancer. 2010;116(17):4160–7. https://doi.org/10.1002/cncr.25427.
Sprague BL, Trentham-Dietz A, Gangnon RE, Ramchandani R, Hampton JM, Robert SA, et al. Socioeconomic status and survival after an invasive breast cancer diagnosis. Cancer. 2011;117(7):1542–51. https://doi.org/10.1002/cncr.25589.
Schwartz KL, Crossley-May H, Vigneau FD, Brown K, Banerjee M. Race, socioeconomic status and stage at diagnosis for five common malignancies. Cancer Causes Control. 2003;14(8):761–6. https://doi.org/10.1023/A:1026321923883.
Lantz PM, Mujahid M, Schwartz K, Janz NK, Fagerlin A, Salem B, et al. The influence of race, ethnicity, and individual socioeconomic factors on breast cancer stage at diagnosis. Am J Public Health. 2006;96(12):2173–8. https://doi.org/10.2105/AJPH.2005.072132.
Huo D, Hu H, Rhie SK, Gamazon ER, Cherniack AD, Liu J, et al. Comparison of breast Cancer molecular features and survival by African and European ancestry in the Cancer genome atlas. JAMA Oncol. 2017;3(12):1654–62. https://doi.org/10.1001/jamaoncol.2017.0595.
Pitt JJ, Riester M, Zheng Y, Yoshimatsu TF, Sanni A, Oluwasola O, et al. Characterization of Nigerian breast cancer reveals prevalent homologous recombination deficiency and aggressive molecular features. Nat Commun. 2018;9(1):4181. https://doi.org/10.1038/s41467-018-06616-0.
Asad S, Barcenas CH, Bleicher RJ, Cohen AL, Javid SH, Levine EG, et al. Abstract P5-07-02: factors associated with rapid relapse in triple negative breast cancer: a multi-institution study. Cancer Res. 2020;80(4 Supplement):P5-07-02-P05-07-02.
Salgado R, Denkert C, Campbell C, Savas P, Nuciforo P, Aura C, et al. Tumor-infiltrating lymphocytes and associations with pathological complete response and event-free survival in HER2-positive early-stage breast Cancer treated with Lapatinib and Trastuzumab: a secondary analysis of the NeoALTTO trial. JAMA Oncol. 2015;1(4):448–54. https://doi.org/10.1001/jamaoncol.2015.0830.
Mao Y, Qu Q, Zhang Y, Liu J, Chen X, Shen K. The value of tumor infiltrating lymphocytes (TILs) for predicting response to neoadjuvant chemotherapy in breast cancer: a systematic review and meta-analysis. PLoS One. 2014;9(12):e115103. https://doi.org/10.1371/journal.pone.0115103.
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28(1):105–13. https://doi.org/10.1200/JCO.2009.23.7370.
Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31(7):860–7. https://doi.org/10.1200/JCO.2011.41.0902.
Telli ML, Stover DG, Loi S, Aparicio S, Carey LA, Domchek SM, et al. Homologous recombination deficiency and host anti-tumor immunity in triple-negative breast cancer. Breast Cancer Res Treat. 2018;171(1):21–31. https://doi.org/10.1007/s10549-018-4807-x.
Adams S, Gatti-Mays ME, Kalinsky K, Korde LA, Sharon E, Amiri-Kordestani L, et al. Current landscape of immunotherapy in breast cancer: a review. JAMA Oncol. 2019;5(8):1205–14. https://doi.org/10.1001/jamaoncol.2018.7147.
Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):e175–86. https://doi.org/10.1016/S1470-2045(19)30026-9.
Pusztai L, Karn T, Safonov A, Abu-Khalaf MM, Bianchini G. New strategies in breast cancer: immunotherapy. Clin Cancer Res. 2016;22(9):2105–10. https://doi.org/10.1158/1078-0432.CCR-15-1315.
Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–90. https://doi.org/10.1038/nrclinonc.2016.66.
Garcia-Murillas I, Chopra N, Comino-Méndez I, Beaney M, Tovey H, Cutts RJ, et al. Assessment of molecular relapse detection in early-stage breast Cancer. JAMA Oncol. 2019;5(10):1473–8. https://doi.org/10.1001/jamaoncol.2019.1838.
Radovich M, Jiang G, Hancock BA, Chitambar C, Nanda R, Falkson C, et al. Association of Circulating Tumor DNA and circulating tumor cells after Neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast Cancer: preplanned secondary analysis of the BRE12-158 randomized clinical trial. JAMA Oncol. 2020;6(9):1410–5. https://doi.org/10.1001/jamaoncol.2020.2295.
Parsons HA, Rhoades J, Reed SC, Gydush G, Ram P, Exman P, et al. Sensitive detection of minimal residual disease in patients treated for early-stage breast Cancer. Clin Cancer Res. 2020;26(11):2556–64. https://doi.org/10.1158/1078-0432.CCR-19-3005.
Hammond MEH, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. Arch Pathol Lab Med. 2010;134(6):907–22. https://doi.org/10.5858/134.6.907.
Allgäuer M, Budczies J, Christopoulos P, Endris V, Lier A, Rempel E, et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics—a primer for molecular pathologists and clinicians. Transl Lung Cancer Res. 2018;7(6):703–15. https://doi.org/10.21037/tlcr.2018.08.14.
Endris V, Buchhalter I, Allgäuer M, Rempel E, Lier A, Volckmar AL, et al. Measurement of tumor mutational burden (TMB) in routine molecular diagnostics: in silico and real-life analysis of three larger gene panels. Int J Cancer. 2019;144(9):2303–12. https://doi.org/10.1002/ijc.32002.
Merino DM, McShane LM, Fabrizio D, Funari V, Chen S-J, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020;8(1):e000147.
Masuda N, Lee SJ, Ohtani S, Im YH, Lee ES, Yokota I, et al. Adjuvant Capecitabine for breast Cancer after preoperative chemotherapy. N Engl J Med. 2017;376(22):2147–59. https://doi.org/10.1056/NEJMoa1612645.
Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, et al. GenVisR: genomic visualizations in R. Bioinformatics. 2016;32(19):3012–4. https://doi.org/10.1093/bioinformatics/btw325.
Acknowledgements
The authors thank Catherine Carson CNP, Celia Garr RN, Ashley Little, and Katherine Weber RN for clinical support making this research possible.
Funding
This research was supported by Susan G. Komen Career Catalyst Research Award (D.G.S.), Pelotonia Young Investigator Award (D.G.S.), and the Stephanie Spielman Fund (D.G.S.).
Author information
Authors and Affiliations
Contributions
Y.Z., W.N., E.P.W., N.U.L, and D.G.S. envisioned the project and designed the work. Y.Z.J., D.M., P.W., L.S., W.H., Z.M.S. generated genomic data for the independent validation dataset. Y.Z., W.N., M.W., Z. W, E.J.A., S.A., D.G.S. wrote the manuscript with input from S.S., D.T., J.T., M.C., M.B.L., B.R., S.S., J.V.D., N.W., AND R.W. All authors have read, revised, and approved the final manuscript. As corresponding author, D.G.S ensures that data, materials, and code comply with transparency and reproducibility standards of the field and journal; ensures that original data/materials/code upon which the submission is based are preserved following best practices in the field so that they are retrievable for reanalysis; confirms that data/materials/code presentation accurately reflects the original; and minimizes obstacles to the sharing of data/materials/code described in the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of The Ohio State University.
Consent for publication
Not Applicable.
Competing interests
E.P.W. has received research grants from Genentech and Roche. N.U.L. has received research grants from Genentech, Array Biopharma, GlaxoSmithKline, Kadmon and Novartis. R.W. has received research support from Acerta and Astra Zeneca and served on advisory boards for PUMA and Pfizer.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
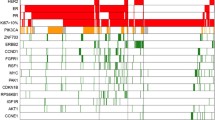
. Additional Analyses of Gene Expression Signatures. (A) Sensitivity analyses of correlation between three representative signatures from each group (immune, proliferation, ER/HER2, mesenchymal) with the immune cell-specific signatures [30, 31] across all samples with gene expression data (n = 453), visualized using CorrPlot [26, 29]. (B) Heatmap with hierarchical clustering of the gene expression signatures with the greatest variance (top 25%) across the dataset. Figure S2. Variation of Expression Signatures Across Rapid vs. Late vs. No Relapse Groups. The calculated score for 16 published gene expression signatures that demonstrated statistical significance (ANOVA FDR p < 0.05) comparing rapid vs. late vs. no relapse. The score value is presented for immune signatures (A) and estrogen/luminal signatures (B). Each boxplot represents the 25th to 75th percentile with the median indicated as the central line and whiskers indicating 1.5 x interquartile range. (C) Immune cell subset proportion from CIBERSORT, visualized as relative values (Z-score) with rapid relapse (red), late relapse (green), and no relapse (blue). Figure S3. Mutation and Modeling Sensitivity Analyses. (A) CoMut plot of gene-level mutation for the entire cohort, with mutation indicated in blue, visualized with ‘GenVisR’ package [68]. (B) Frequency of gene-level copy number gains (red) or losses (blue) across the genome
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Asad, S., Weber, Z. et al. Genomic features of rapid versus late relapse in triple negative breast cancer. BMC Cancer 21, 568 (2021). https://doi.org/10.1186/s12885-021-08320-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08320-7