
Abstract
Background
Lung adenocarcinoma (LAC) is the predominant histologic subtype of lung cancer and has a complicated pathogenesis with high mortality. The purpose of this study was to identify differentially expressed genes (DEGs) with prognostic value and determine their underlying mechanisms.
Methods
Gene expression data of GSE27262 and GSE118370 were acquired from the Gene Expression Omnibus database, enrolling 31 LAC and 31 normal tissues. Common DEGs between LAC and normal tissues were identified using the GEO2R tool and Venn diagram software. Next, the Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used to analyze the Gene Ontology and Kyoto Encyclopedia of Gene and Genome (KEGG) pathways. Then, protein-protein interaction (PPI) network of DEGs was visualized by Cytoscape with Search Tool for the Retrieval of Interacting Genes and central genes were identified via Molecular Complex Detection. Furthermore, the expression and prognostic information of central genes were validated via Gene Expression Profiling Interactive Analysis (GEPIA) and Kaplan-Meier analysis, respectively. Finally, DAVID, real-time PCR and immunohistochemistry were applied to re-analyze the identified genes, which were also further validated in two additional datasets from ArrayExpress database.
Results
First, 189 common DEGs were identified among the two datasets, including 162 downregulated and 27 upregulated genes. Next, Gene Ontology and KEGG pathway analysis of the DEGs were conducted through DAVID. Then, PPI network of DEGs was constructed and 17 downregulated central genes were identified. Furthermore, the 17 downregulated central genes were validated via GEPIA and datasets from ArrayExpress, and 12 of them showed a significantly better prognosis. Finally, six genes were identified significantly enriched in neuroactive ligand-receptor interactions (EDNRB, RXFP1, P2RY1, CALCRL) and Rap1 signaling pathway (TEK, P2RY1, ANGPT1) via DAVID, which were further validated to be weakly expressed in LAC tissues via RNA quantification and immunohistochemistry analysis.
Conclusions
The low expression pattern and relation to prognosis indicated that the six genes were potential tumor suppressor genes in LAC. In conclusion, we identified six significantly downregulated DEGs as prognostic markers and potential tumor suppressor genes in LAC based on integrated bioinformatics methods, which could act as potential molecular markers and therapeutic targets for LAC patients.
Similar content being viewed by others


Background
Lung cancer remains the leading cause of cancer-related deaths in men and women worldwide [1, 2]. In China, both incidence and mortality from lung cancer continue to increase, which poses a significant threat to public health [3]. The complicated pathogenesis of lung cancer result from a variety of risk factors, most commonly include lifestyle, environmental, occupational exposure, and genetic factors [1]. Adenocarcinoma is the predominant histologic subtype of lung cancer both in men and women [1, 4]. However, despite advances in tumor biology and treatment, the five-year overall survival rate is approximately 15.7% [5], and varies markedly depending on the stage when the diagnosis is made [6]. Thus, it is essential to identify specific molecular markers and develop a more personalized therapy for lung adenocarcinoma (LAC) to improve early prediction and outcomes.
To date, molecular markers have been widely studied for the detection and prognosis of LAC. Runt-related transcription factor 3 (RUNX3) [7], estrogen receptor [8], and chemokine receptor [9, 10] have been identified as good prognostic markers, whose high expression is significantly correlated with an increase in disease-free survival in LAC patients. In addition, Ets-1 [11], Kruppel-like factor 6 (KLF6) [12], eukaryotic initiation factor 4E (eIF4E) [13], Nectin-like molecule-5 (Necl-5) [14], and histone deacetylases (HDACs) [15, 16] have been demonstrated as poor prognostic markers [17]. Furthermore, molecular markers have been tested as targets of specific therapies for LAC patients. Epidermal growth factor receptor (EGFR) insertions and deletions have been found in approximately 15% of LAC patients in the United States [18], which indicates a favorable sensitivity to tyrosine kinase inhibitors towards EGFR [19]. KRAS mutations have been commonly found in smokers and appear to confer a worse prognosis [18]. Drugs that target KRAS mutations are actively testing in clinical trials [19]. Additional gene mutations, such as BRAF mutations, HER2 mutations, ROS1 translocations, and ALK gene rearrangements, could also be targets in LAC patients.
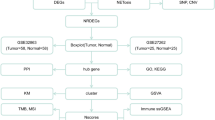
However, an increase in molecular markers for lung adenocarcinoma is still in urgent demand. The datasets of gene expression profiles in the Gene Expression Omnibus (GEO) are far from being excavated and contain a great deal of information regarding LAC. The bioinformatic analysis provides a powerful and comprehensive tool for analyzing gene expression data from multiple datasets. Thus, in this study, we first searched the gene expression profiling datasets of LAC in GEO and finally chose GSE27262 and GSE118370 for bioinformatic analysis. Second, we applied the GEO2R and Venn diagram software to identify the common differentially expressed genes (DEGs) between the two datasets. Then, Gene Ontology and pathway enrichment were analyzed through the Database for Annotation, Visualization and Integrated Discovery (DAVID), including the molecular function (MF), cellular component (CC), biological process (BP), and Kyoto Encyclopedia of Gene and Genome (KEGG) pathways [20]. Furthermore, we constructed a protein-protein interaction (PPI) network and then applied the Cytoscape Molecular Complex Detection (MCODE) to identify the core genes in the PPI network. Moreover, we validated the core gene’s expression between LAC tissues and normal lung tissues via Gene Expression Profiling Interactive Analysis (GEPIA) and ArrayExpress datasets. In addition, these core genes were further analyzed for significant prognostic information based on the Kaplan-Meier online database. Thus, 12 core genes were qualified and KEGG pathway enrichment was re-analyzed. Finally, six genes were generated, which were mainly enriched in neuroactive ligand-receptor interactions and Rap1 signaling pathway, and their further expressions were validated via RNA quantification and immunohistochemistry analysis in tissue samples. The low expression of the six genes and their relation to prognosis in LAC indicated that they were potential tumor suppressor genes. In conclusion, our bioinformatics study identified useful and potential tumor suppressor genes that could potentially act as biomarkers and effective targets for LAC patients.
Methods
Microarray data information
NCBI-GEO is a widely used public database and provides gene expression profile of numerous cancers for study. The keywords in the search process were as follows: lung cancer, non-small cell lung cancer, lung adenocarcinoma, and GPL570. The following criteria were used to screen the datasets and ensure relevant data were recorded: (I) the sample includes lung adenocarcinoma and paired adjacent tissues; (II) the study type is expression profiling by array; (III) the species is limited to Homo sapiens; (IV) access to raw data is allowed. We obtained the gene expression data of GSE27262 and GSE118370 in lung adenocarcinoma and paired normal lung tissues for bioinformatics analysis. Microarray data of GSE27262 and GSE118370 were based on GPL570 Platforms ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array), including 25 LAC tissues and 25 paired normal lung tissues, 6 LAC tissues and 6 paired normal lung tissues, respectively. Another two datasets from ArrayExpress database, E-GEOD-30219 and E-GEOD-19188, which include specimens from LAC of variable TNM stages, were enrolled to further validate the expression of identified genes.
Data processing of DEGs
Robust multi-array average (RMA) and MicroArray Suite (MAS) approach was performed for background correction and normalization. The GEO2R online tools [21] on the NCBI-GEO website, which using the GEO query and limma R packages to analyze high-throughput genomic data, were used to identify DEGs between the LAC specimen and normal lung specimen with |log2FC| > 2 and an adjusted P value < 0.05 [20]. Then, the raw data were analyzed using Venn software online to identify the common DEGs among the original two datasets. DEGs with log2FC < 0 were considered as downregulated genes, while the DEGs with log2FC > 0 were considered as upregulated genes [20].
Gene ontology and KEGG pathway enrichment analysis
DAVID [22] is an online bioinformatics tool that integrates the function of Gene Ontology and KEGG pathway enrichment analysis [23, 24]. Through DAVID, we identified the unique biological properties of the common DEGs and visualized the DEGs enrichment of molecular function (MF), cellular components (CC), biological processes (BP), and KEGG pathways (P < 0.05).
PPI network construction and module analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) online tool [25] was used to evaluate PPI information. Thereafter, Cytoscape [26] was used to visualize the potential correlation and interaction between these DEGs (maximum number of interactors = 0 and confidence score ≥ 0.4). In addition, the MCODE app was applied to inspect the central modules of the PPI network (degree cutoff = 2, max. Depth = 100, k-core = 2, and node score cutoff = 0.2) [20]. PPI network properties, such as node degree and betweenness centrality, were visualized by shape size and label font size, respectively.
Survival and RNA sequencing analysis
The Kaplan-Meier plotter is a useful website tool, which contains considerable information on several cancers, including breast and lung cancer [27]. Survival analysis was conducted using the Kaplan-Meier plotter, and the log-rank P value and hazard ratio (HR) with 95% confidence intervals were computed and shown on the plot [20]. To further validate these DEGs with significant survival outcomes, we applied the Gene Expression Profiling Interactive Analysis (GEPIA) website to analyze the RNA sequencing data based on the GTEx projects and TCGA database [28].
RNA quantification
Total RNA was extracted with Trizol reagent (Invitrogen) and reverse-transcribed using the PrimeScript™ RT reagent kit (Takara). Quantitative real-time PCR analysis was performed on a LightCycler (Roche) with the TB Green® Premix Ex Taq™ II (Takara). Data were normalized to GAPDH expression. The primers used for real-time PCR were as follows: GAPDH (forward: 5′-GGA GCG AGA TCC CTC CAA AAT-3′, reverse: 5′-GGC TGT TGT CAT ACT TCT CAT GG-3′), EDNRB (forward: 5′-CTG GCC ATT TGG AGC TGA GA-3′, reverse: 5′-CAG AAC CAC AGA GAC CAC CC-3′), RXFP1 (forward: 5′-GGA CCT GAA GGA GCT GTC AC-3′, reverse: 5′-AGG CTG AGA GAC TTG AGT TTG A-3′), P2RY1 (forward: 5′-CCG TCT CCT CGT CGT TCA AA-3′, reverse: 5′-ACG TAC AAG AAG TCG GCC AG-3′), CALCRL (forward: 5′-CCC ACC TTG CTT GTG GGT AA-3′, reverse: 5′-GTC AAG ACC CAG TCC AGC TC-3′), TEK (forward: 5′-CCA GCC CTG CTG ATA CCA AA-3′, reverse: 5′-AGG CAA GAA GGA ACA GCA CA-3′), and ANGPT1 (forward: 5′-TCC AGG AGC TGG AAA AGC AA-3′, reverse: 5′-TGC AAA GAT TGA CAA GGT TGT GG-3′).
Immunohistochemical (IHC) staining
IHC staining was applied to detect the protein level of certain genes and performed according to standard protocols using the following antibodies: anti-EDNRB (bs-2363R, 1:500), anti-RXFP1 (bs-15368R, 1:500), anti-P2RY1 (EM1710–48, 1:200), anti-CALCRL (BA1572–1, 1:200), anti-TEK (bs-1300R, 1:500), and anti-ANGPT1 (bs-0800R, 1:500).
Results
Identification of DEGs in lung adenocarcinoma
In the present study, the gene expression data of GSE27262 and GSE118370 were chosen for bioinformatic analysis, including 31 LAC tissues and 31 normal lung tissues. Using the GEO2R online tool, we obtained 474 and 409 DEGs from GSE27262 and GSE118370 (Fig. 1a & b), respectively (|log2FC| > 2 and adjusted P value < 0.05). Then, we applied Venn diagram software to identify the common DEGs among the two datasets. Results showed that 189 common DEGs were identified, including 162 downregulated genes (log2FC < 0) and 27 upregulated genes (log2FC > 0) in LAC tissues (Table 1, S1 & S2, and Fig. 1c & d).

Identification of 189 common DEGs among GSE27262 and GSE118370 datasets by Venn diagram software. Different colors represent different datasets. a, c 162 DEGs were downregulated among the two datasets (log2FC < −2). b, d 27 DEGs were upregulated among the two datasets (log2FC > 2)
Gene ontology and KEGG analysis of DEGs in lung adenocarcinoma
To examine the biological properties of the 189 DEGs, Gene Ontology and KEGG analysis were conducted via DAVID software. Results of Gene Ontology analysis indicated that 1) for biological processes (BP), downregulated DEGs were significantly enriched in angiogenesis, vasculogenesis signal transduction, receptor internalization, and neural crest cell migration, and upregulated DEGs were enriched in collagen catabolic processes, proteolysis, sensory perception of sound, and embryonic cranial skeleton morphogenesis; 2) for cell components (CC), downregulated DEGs were significantly enriched in the integral components of membrane, plasma membrane, proteinaceous extracellular matrix and cell-cell junctions, and upregulated DEGs were significantly enriched in extracellular space, extracellular regions, extracellular exosomes, and the proteinaceous extracellular matrix; 3) for molecular function (MF), downregulated DEGs were enriched in semaphoring receptor binding, chemorepellent activity, heparin-binding, and calcium ion-binding, and upregulated DEGs were enriched in endopeptidase inhibitor activity, serine-type endopeptidase inhibitor activity (P < 0.05, Table 2).
The results of KEGG analysis showed that downregulated DEGs were particularly enriched in neuroactive ligand-receptor interactions, cell adhesion molecules, axon guidance, hypertrophic cardiomyopathy, and vascular smooth muscle contractions, while upregulated DEGs were not enriched in any significant signaling pathways (P < 0.05, Table 3).
The PPI network construction and modular analysis of DEGs
To analyze the PPI information of the 189 DEGs, STRING online database and Cytoscape software were used to construct the PPI network complex. In total, 137 DEGs were enrolled in the PPI network, which included 137 nodes and 254 edges, including 122 downregulated and 15 upregulated genes (Fig. 2a and Table S3). There were 52 DEGs not presented in the DEGs PPI network. We then used Cytoscape MCODE to further screen the core genes, and results revealed that 17 central nodes, all of which were downregulated, were identified (Fig. 2b and Table S3).

Common DEGs PPI network was constructed using the STRING online database and Cytoscape software. a There were 137 nodes and 254 edges in the PPI network. The nodes represent proteins; the edges represent the interaction between proteins; green circles represent downregulated DEGs, and red circles represent upregulated DEGs. b Modular analysis via MCODE (degree cutoff = 2, max. Depth = 100, k-core = 2, and node score cutoff = 0.2). In total, 17 central nodes were screened. Circle size represents node degree, and label font size represents betweenness centrality
Analysis of 17 core genes via the GEPIA and Kaplan-Meier plotter
To further validate the significance of the 17 central genes, GEPIA and the Kaplan-Meier plotter were utilized to identify the expression data and survival data, respectively. GEPIA expression data showed that all 17 genes were lowly expressed in LAC tissues compared to normal lung tissues (P < 0.05, Table 4 and Fig. 3). The Kaplan-Meier plotter survival data showed that a high expression of 12 of the 17 genes resulted in a significantly better survival probability, while high expression of ADRA1A, TIE1, and LYVE1 had a significantly worse survival probability; VIPR1 and RAMP3 were not significantly different (P < 0.05, Table 5 and Fig. 4).

Expression levels of the 17 central genes in lung adenocarcinoma patients compared to healthy people. The GEPIA website was applied to validate the expression level of the 17 central genes between LAC patients and normal people. All 17 genes were lowly expressed in LAC specimens compared to normal specimens (*P < 0.05). Red indicates LAC tissues (n = 483) and gray indicates normal tissues (n = 347)

Prognostic information of the 17 central genes in lung adenocarcinoma. The Kaplan-Meier plotter online tools were used to analyze the prognostic information of the 17 central genes. a High expression of 12 of the 17 genes had a significantly better survival rate (P < 0.05). b High expression of ADRA1A, TIE1, and LYVE1 showed a significantly worse survival rate (P < 0.05)
Re-analysis of 12 core genes by KEGG, RNA quantification and immunohistochemistry
To figure out the possible pathway of the 12 core DEGs, DAVID was used for KEGG pathway enrichment analysis. The results showed that six core genes were markedly enriched, in which four genes (EDNRB, RXFP1, P2RY1, and CALCRL) were enriched in neuroactive ligand-receptor interactions, and three genes (TEK, P2RY1, and ANGPT1) were enriched in the Rap1 signaling pathway (P < 0.05, Table 6 and Figs. 5 & 6).

General information of neuroactive ligand-receptor interactions pathway. DAVID was used to re-analyze the 12 core DEGs for KEGG pathway enrichment. Four genes (EDNRB, RXFP1, P2RY1, and CALCRL) were enriched in neuroactive ligand-receptor interactions (P < 0.05)

General information of Rap1 signaling pathway. DAVID was used to re-analyze the 12 core DEGs for KEGG pathway enrichment. Three genes (TEK, P2RY1, and ANGPT1) were enriched in Rap1 signaling pathway (P < 0.05)
Furthermore, we detected the expression levels of the above six genes in LAC specimens and normal lung specimens by RNA quantification and immunohistochemistry analysis. Results showed that all six genes were lowly expressed in LAC tissues compared to adjacent normal tissues (Fig. 7).

Validation of expression levels of EDNRB, RXFP1, P2RY1, CALCRL, TEK, and ANGPT1 in LAC patients. To further validate the expression level in LAC patients, six genes were re-analyzed via real-time PCR (a) and immunohistochemistry (b) analysis. Representative images of IHC staining were shown. Scale bar, 50 μm. Real-time PCR data were presented as mean ± SEM and the differences were estimated by Wilcoxon paired signed-rank test (*P < 0.05, **P < 0.01, ***P < 0.001). Data were normalized to GAPDH expression. All six genes were markedly weakly expressed in LAC tissue compared to adjacent normal tissue
Finally, we further validated the expression of identified common DEGs, especially the 17 central genes in E-GEOD-30219 and E-GEOD-19188 datasets. The validated datasets were acquired from ArrayExpress database and included specimens from lung adenocarcinoma of variable TNM stage. Results showed that the vast majority of the previously identified 189 common DEGs and all 17 central genes were included in the DEGs of the two datasets with same expression pattern (Table S4 & S5).
Discussion
In this study, we applied bioinformatics methods based on two gene expression profile datasets to identify additional useful prognostic molecular markers in lung adenocarcinoma. Thirty-one LAC specimens and thirty-one paired normal lung specimens were enrolled. Using GEO2R online tool and Venn software, we revealed 189 common DEGs (|log2FC| > 2 and adjusted P value < 0.05), including 162 downregulated and 27 upregulated DEGs. Gene Ontology and KEGG pathway enrichment analysis was conducted via DAVID. Gene Ontology analysis revealed that 1) for biological processes, downregulated DEGs were particularly enriched in angiogenesis, vasculogenesis signal transduction, receptor internalization, and neural crest cell migration, and upregulated DEGs were enriched in collagen catabolic process, proteolysis, sensory perception of sound, and embryonic cranial skeleton morphogenesis; 2) for cell components, downregulated DEGs were significantly enriched in integral components of the membrane, plasma membrane, proteinaceous extracellular matrix and cell-cell junctions, and upregulated DEGs were significantly enriched in the extracellular space, extracellular region, extracellular exosome, and proteinaceous extracellular matrix; 3) for molecular function, downregulated DEGs were significantly enriched in semaphoring receptor binding, chemorepellent activity, heparin-binding, and calcium ion-binding, and upregulated DEGs were enriched in endopeptidase inhibitor activity and serine-type endopeptidase inhibitor activity (P < 0.05). For pathway analysis, downregulated DEGs were particularly enriched in neuroactive ligand-receptor interactions, cell adhesion molecules, axon guidance, hypertrophic cardiomyopathy, and vascular smooth muscle contractions, while upregulated DEGs were not enriched in any significant signaling pathways (P < 0.05). Next, the PPI network complex of 137 nodes and 254 edges was constructed using the STRING and Cytoscape software. Thereafter, 17 central downregulated DEGs were screened from the PPI network by MCODE analysis. In addition, GEPIA analysis showed that all 17 genes were lowly expressed in LAC tissues (P < 0.05). Furthermore, through Kaplan-Meier analysis, we found that a high expression of 12 of the 17 genes displayed increased survival. Finally, we re-analyzed the 12 core genes via DAVID, RNA quantification, and immunohistochemistry. Six genes were markedly enriched and downregulated in LAC samples, in which four genes (EDNRB, RXFP1, P2RY1, and CALCRL) were enriched in neuroactive ligand-receptor interactions, and three genes (TEK, P2RY1, and ANGPT1) were enriched in the Rap1 signaling pathway (P < 0.05). Altogether, we identified six significant genes with good prognosis and tumor suppressor function in lung adenocarcinoma via bioinformatics analysis, which could be new molecular markers and effective targets for further research.
There was some evidence that EDNRB, RXFP1, ANGPT1, and TEK are closely related to lung diseases and cancer. For example, promoter hypermethylation of the EDNRB gene was found in several human tumors, including lung cancer. The endothelin receptor type B (EDNRB) gene encodes a G-protein coupled receptor and is regulated by the methylation of its CpG sites [29, 30]. Aberrant methylation of the EDNRB gene was detected in 32.9% (26 of 79) of lung cancer patients, which then decreased EDNRB expression and contributed to tumor progression [31]. These findings show that aberrant methylation of the EDNRB gene is highly prevalent in lung cancer. A previous study also identified EDNRB as a potential molecular marker for LAC via integrated bioinformatic analysis [17]. Recently, it was reported that EDNRB expression was significantly increased in chronic obstructive pulmonary disease (COPD) patients and was effectively reduced after celastrol treatment, which supposes an inflammation-related role of EDNRB in COPD [32].
Relaxin family peptide receptor-1 (RXFP1), also known as LGR7, is a leucine-rich repeat that contains a G-protein coupled receptor and is expressed in human and mouse lungs [33]. Evidence shows that RXFP1 appears to have a significant impact on lung diseases. A previous study revealed that Rxfp1-deficient mice had increased lung collagen accumulation as early as 1 month of age [34], indicating that RXFP1 could delay the age-related progression of pulmonary fibrosis. Further studies have demonstrated that RXFP1 protects against airway fibrosis during homeostasis but not against inflammation-induced fibrosis associated with chronic allergic airways [35]. More importantly, RXFP1 expression was reported to be directly associated with pulmonary function in patients with idiopathic pulmonary fibrosis (IPF), and results showed that patients with IPF and high RXFP1 expression are more sensitive to relaxin-based therapies [36].
Angiopoietin-1 (ANGPT1), a secreted glycoprotein, is a physiological angiogenesis promoter during embryonic development and has an enigmatic role in tumor angiogenesis [37]. The TEK receptor tyrosine kinase (TEK), also known as TIE2, is a receptor for ANGPT1 and belongs to the protein tyrosine kinase Tie2 family. Physiologically, ANGPT1 binds to TEK to mediate embryonic vascular development and angiogenesis [37, 38]. A previous study showed that normal lung tissues expressed constitutively high and correlated levels of ANGPT1 and TEK, which were significantly reduced in non-small cell lung cancers (NSCLC) [39]. These previous findings indicated a specified role of the ANGPT1/TEK pathway in the maintenance of the complex vasculature in normal lungs. Evidence has shown that lung cancer patients with a higher level of ANGPT1 had better survival, indicating that ANGPT1 is a prognostic marker for lung cancer, especially for predicting postoperative survival and recurrence [40]. Furthermore, a recent study demonstrated that ANGPT1 could be a potential tumor suppressor gene for lung cancer [41]. Alterations in the intron region of ANGPT1 were found in lung cancer and affected the expression level of ANGPT1, which lead to the neoplastic progression of lung cancer. Moreover, survival analysis found that high expression of ANGPT1 associated with a higher survival probability individually [41].
Although there have been no reports of P2RY1 and CALCRL involved in lung diseases and cancer, they all have been demonstrated to play important roles in other cancers, such as bladder cancer [42], prostate cancer [43], and acute myeloid leukemia [44]. They are prognostic markers or key molecules for tumorigenesis in these cancers. Therefore, they may also be potential markers for LAC and require further study. In short, our study provides some useful information and clues for future studies in LAC.
Conclusions
Our bioinformatic analysis identified six downregulated DEGs (EDNRB, RXFP1, P2RY1, CALCRL, TEK, and ANGPT1) between lung adenocarcinoma and normal lung tissues based on two different microarray datasets. These six genes were identified as excellent prognostic markers and potential tumor suppressors, playing key roles in the initiation and progression of LAC. However, more studies are required to verify the prediction and underlying mechanisms in the near future. These data may provide novel perspectives and clues into the study of potential molecular markers and therapeutic targets in LAC.
Availability of data and materials
Dataset supporting our findings is available, at the following website: www.ncbi.nlm.nih.gov/geo/, www.ebi.ac.uk/arrayexpress/. All data generated or analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- LAC:
-
Lung adenocarcinoma
- GEO:
-
Gene Expression Omnibus
- DEGs:
-
Differentially expressed genes
- EDNRB:
-
Endothelin receptor type B
- RXFP1:
-
Relaxin family peptide receptor-1
- P2RY1:
-
Purinergic receptor P2Y1
- CALCRL:
-
Calcitonin receptor like receptor
- TEK:
-
TEK receptor tyrosine kinase
- ANGPT1:
-
Angiopoietin-1
- PPI:
-
Protein-protein interaction
- DAVID:
-
The Database for Annotation, Visualization and Integrated Discovery
- GO:
-
Gene ontology
- KEGG pathways:
-
Kyoto Encyclopedia of Gene and Genome pathways
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
- MCODE:
-
Molecular Complex Detection
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- TCGA:
-
The Cancer Genome Atlas
References
Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung Cancer. Ann Global Health. 2019;85(1):8. https://doi.org/10.5334/aogh.2419.
Liu B, Quan X, Xu C, Lv J, Li C, Dong L, et al. Lung cancer in young adults aged 35 years or younger: a full-scale analysis and review. J Cancer. 2019;10(15):3553–9. https://doi.org/10.7150/jca.27490.
Li J, Yang F, Li X, Zhang M, Fu R, Yin X, et al. Characteristics, survival, and risk factors of Chinese young lung cancer patients: the experience from two institutions. Oncotarget. 2017;8(51):89236–44. https://doi.org/10.18632/oncotarget.19183.
Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–85. https://doi.org/10.1097/JTO.0b013e318206a221.
Barletta JA, Yeap BY, Chirieac LR. Prognostic significance of grading in lung adenocarcinoma. Cancer. 2010;116(3):659–69. https://doi.org/10.1002/cncr.24831.
Alberg AJ, Brock MV, Ford JG, Samet JM, Spivack SD. Epidemiology of lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):e1S–e29S. https://doi.org/10.1378/chest.12-2345.
Araki K, Osaki M, Nagahama Y, Hiramatsu T, Nakamura H, Ohgi S, et al. Expression of RUNX3 protein in human lung adenocarcinoma: implications for tumor progression and prognosis. Cancer Sci. 2005;96(4):227–31. https://doi.org/10.1111/j.1349-7006.2005.00033.x.
Nose N, Sugio K, Oyama T, Nozoe T, Uramoto H, Iwata T, et al. Association between estrogen receptor-beta expression and epidermal growth factor receptor mutation in the postoperative prognosis of adenocarcinoma of the lung. J Clin Oncol. 2009;27(3):411–7. https://doi.org/10.1200/JCO.2008.18.3251.
Minamiya Y, Saito H, Takahashi N, Ito M, Imai K, Ono T, et al. Expression of the chemokine receptor CXCR4 correlates with a favorable prognosis in patients with adenocarcinoma of the lung. Lung Cancer. 2010;68(3):466–71. https://doi.org/10.1016/j.lungcan.2009.07.015.
Minamiya Y, Saito H, Takahashi N, Ito M, Toda H, Ono T, et al. Expression of the chemokine receptor CCR6 correlates with a favorable prognosis in patients with adenocarcinoma of the lung. Tumour Biol. 2011;32(1):197–202. https://doi.org/10.1007/s13277-010-0113-x.
Yamaguchi E, Nakayama T, Nanashima A, Matsumoto K, Yasutake T, Sekine I, et al. Ets-1 proto-oncogene as a potential predictor for poor prognosis of lung adenocarcinoma. Tohoku J Exp Med. 2007;213(1):41–50. https://doi.org/10.1620/tjem.213.41.
DiFeo A, Feld L, Rodriguez E, Wang C, Beer DG, Martignetti JA, et al. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008;68(4):965–70. https://doi.org/10.1158/0008-5472.CAN-07-2604.
Wang R, Geng J, Wang JH, Chu XY, Geng HC, Chen LB. Overexpression of eukaryotic initiation factor 4E (eIF4E) and its clinical significance in lung adenocarcinoma. Lung Cancer. 2009;66(2):237–44. https://doi.org/10.1016/j.lungcan.2009.02.001.
Nakai R, Maniwa Y, Tanaka Y, Nishio W, Yoshimura M, Okita Y, et al. Overexpression of Necl-5 correlates with unfavorable prognosis in patients with lung adenocarcinoma. Cancer Sci. 2010;101(5):1326–30. https://doi.org/10.1111/j.1349-7006.2010.01530.x.
Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, et al. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer. 2011;74(2):300–4. https://doi.org/10.1016/j.lungcan.2011.02.019.
Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Motoyama S, et al. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumour Biol. 2010;31(5):533–9. https://doi.org/10.1007/s13277-010-0066-0.
Wu X, Zang W, Cui S, Wang M. Bioinformatics analysis of two microarray gene-expression data sets to select lung adenocarcinoma marker genes. Eur Rev Med Pharmacol Sci. 2012;16(11):1582–7.
Dogan S, Shen R, Ang DC, Johnson ML, D'Angelo SP, Paik PK, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res. 2012;18(22):6169–77. https://doi.org/10.1158/1078-0432.CCR-11-3265.
Ou SH. Lung cancer in never-smokers. Does smoking history matter in the era of molecular diagnostics and targeted therapy? J Clin Pathol. 2013;66(10):839–46. https://doi.org/10.1136/jclinpath-2012-201296.
Feng H, Gu Z-Y, Li Q, Liu Q-H, Yang X-Y, Zhang J-J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J Ovar Res. 2019;12(1):35. https://doi.org/10.1186/s13048-019-0508-2.
Davis S, Meltzer PS. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–7. https://doi.org/10.1093/bioinformatics/btm254.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. https://doi.org/10.1038/75556.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. https://doi.org/10.1093/nar/28.1.27.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52. https://doi.org/10.1093/nar/gku1003.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8(1):9227. https://doi.org/10.1038/s41598-018-27521-y.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. https://doi.org/10.1093/nar/gkx247.
Puffenberger EG, Hosoda K, Washington SS, Nakao K, de Wit D, Yanagisawa M, et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell. 1994;79(7):1257–66. https://doi.org/10.1016/0092-8674(94)90016-7.
Eberle J, Weitmann S, Thieck O, Pech H, Paul M, Orfanos CE. Downregulation of endothelin B receptor in human melanoma cell lines parallel to differentiation genes. J Invest Dermatol. 1999;112(6):925–32. https://doi.org/10.1046/j.1523-1747.1999.00598.x.
Chen SC, Lin CY, Chen YH, Fang HY, Cheng CY, Chang CW, et al. Aberrant promoter methylation of EDNRB in lung cancer in Taiwan. Oncol Rep. 2006;15(1):167–72.
Shi K, Chen X, Xie B, Yang SS, Liu D, Dai G, et al. Celastrol alleviates chronic obstructive pulmonary disease by inhibiting cellular inflammation induced by cigarette smoke via the Ednrb/Kng1 signaling pathway. Front Pharmacol. 2018;9:1276. https://doi.org/10.3389/fphar.2018.01276.
Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD, et al. Activation of orphan receptors by the hormone relaxin. Science. 2002;295(5555):671–4. https://doi.org/10.1126/science.1065654.
Feng S, Bogatcheva NV, Kamat AA, Agoulnik AI. Genetic targeting of relaxin and insl3 signaling in mice. Ann N Y Acad Sci. 2005;1041(1):82–90. https://doi.org/10.1196/annals.1282.012.
Samuel CS, Royce SG, Chen B, Cao H, Gossen JA, Tregear GW, et al. Relaxin family peptide receptor-1 protects against airway fibrosis during homeostasis but not against fibrosis associated with chronic allergic airways disease. Endocrinology. 2009;150(3):1495–502. https://doi.org/10.1210/en.2008-1062.
Tan J, Tedrow JR, Dutta JA, Juan-Guardela B, Nouraie M, Chu Y, et al. Expression of RXFP1 is decreased in idiopathic pulmonary fibrosis. Implications for Relaxin-based therapies. Am J Respir Crit Care Med. 2016;194(11):1392–402. https://doi.org/10.1164/rccm.201509-1865OC.
Metheny-Barlow LJ, Li LY. The enigmatic role of angiopoietin-1 in tumor angiogenesis. Cell Res. 2003;13(5):309–17. https://doi.org/10.1038/sj.cr.7290176.
Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87(7):1171–80. https://doi.org/10.1016/S0092-8674(00)81813-9.
Wong MP, Chan SY, Fu KH, Leung SY, Cheung N, Yuen ST, et al. The angiopoietins, tie2 and vascular endothelial growth factor are differentially expressed in the transformation of normal lung to non-small cell lung carcinomas. Lung Cancer. 2000;29(1):11–22. https://doi.org/10.1016/S0169-5002(00)00118-5.
Park JH, Choi H, Kim YB, Kim YS, Sheen SS, Choi JH, et al. Serum angiopoietin-1 as a prognostic marker in resected early stage lung cancer. Lung Cancer. 2009;66(3):359–64. https://doi.org/10.1016/j.lungcan.2009.03.002.
Yao S, Dong SS, Ding JM, Rong Y, Zhang YJ, Chen H, et al. Sex-specific SNP-SNP interaction analyses within topologically associated domains reveals ANGPT1 as a novel tumor suppressor gene for lung cancer. Genes Chromosom Cancer. 2019;59(1):13–22. https://doi.org/10.1002/gcc.22793.
Tan Y, Zhang T, Zhou L, Liu S, Liang C. MiR-34b-3p represses the multidrug-Chemoresistance of bladder Cancer cells by regulating the CCND2 and P2RY1 genes. Med Sci Monit:1323–35. https://doi.org/10.12659/MSM.913746.
Light KC, Agarwal N, Iacob E, White AT, Kinney AY, VanHaitsma TA, et al. Differing leukocyte gene expression profiles associated with fatigue in patients with prostate cancer versus chronic fatigue syndrome. Psychoneuroendocrinology. 2013;38(12):2983–95. https://doi.org/10.1016/j.psyneuen.2013.08.008.
Angenendt L, Bormann E, Pabst C, Alla V, Gorlich D, Braun L, et al. The neuropeptide receptor calcitonin receptor-like (CALCRL) is a potential therapeutic target in acute myeloid leukemia. Leukemia. 2019;33(12):2830–41. https://doi.org/10.1038/s41375-019-0505-x.
Acknowledgements
We thank Ms. Yamei Li and Dr. Yong Zhang for technical assistance and language help.
Funding
This study was supported by grants from the National Natural Science Foundation of China (grant no.81702287, 81700766).
Author information
Authors and Affiliations
Contributions
HZ and CW designed the study; MZL, XWF, WLL, YJL, LJM, MY, RG, ZDW acquired and analyzed the data; MZL, XWF, WLL, CW and HZ wrote the paper. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of the General Hospital of Western Theater Command and informed consent was exempted.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
General information of the differentially expressed genes in GSE27262 datasets. Table S1–1. Down-regulated genes in GSE27262 datasets. Table S1–2. Up-regulated genes in GSE27262 datasets.
Additional file 2: Table S2.
General information of the differentially expressed genes in GSE118370 datasets. Table S2–1. Down-regulated genes in GSE118370 datasets. Table S2–2. Up-regulated genes in GSE118370 datasets.
Additional file 4: Table S4.
Validation of the 189 common DEGs in E-GEOD-30219 datasets. Table S4–1. Validation of the 162 common down-regulated DEGs in E-GEOD-30219 datasets. Table S4–2. Validation of the 27 common up-regulated DEGs in E-GEOD-30219 datasets.
Additional file 5: Table S5.
Validation of the 189 common DEGs in E-GEOD-19188 datasets. Table S5–1. Validation of the 162 common down-regulated DEGs in E-GEOD-19188 datasets. Table S5–2. Validation of the 27 common up-regulated DEGs in E-GEOD-19188 datasets.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, M., Fan, X., Liao, W. et al. Identification of significant genes as prognostic markers and potential tumor suppressors in lung adenocarcinoma via bioinformatical analysis. BMC Cancer 21, 616 (2021). https://doi.org/10.1186/s12885-021-08308-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08308-3