
Abstract
Background
Intrahepatic cholestasis of pregnancy (ICP) is characterized by pruritus and cholestasis in late pregnancy and results in adverse pregnancy outcomes, including preterm delivery and birth weight, which are affected by the genetic and environmental background. However, until now, the genetic architecture of ICP has remained largely unclear.
Methods
Twenty-six clinical data points were recorded for 151 Chinese ICP patients. The data generated from whole-exome sequencing (WES) using the BGISEQ-500 platform were further analyzed by Burrows-Wheeler Aligner (BWA) software, Genome Analysis Toolkit (GATK), ANNOVAR tool, etc. R packages were used to conduct t-test, Fisher’s test and receiver operating characteristic (ROC) curve analyses.
Results
We identified eighteen possible pathogenic loci associated with ICP disease in known genes, covering ABCB4, ABCB11, ATP8B1 and TJP2. The loci Lys386Gln, Gly527Gln and Trp708Ter in ABCB4, Leu589Met, Gln605Pro and Gln1194Ter in ABCB11, and Arg189Ser in TJP2 were novel discoveries. In addition, WES analysis indicated that the gene ANO8 involved in the transport of bile salts is newly identified as associated with ICP. The functional network of the ANO8 gene confirmed this finding. ANO8 contained 8 rare missense mutations that were found in eight patients among the 151 cases and were absent from 1029 controls. Out of the eight SNPs, 3 were known, and the remaining five are newly identified. These variants have a low frequency, ranging from 0.000008 to 0.00001 in the ExAC, gnomAD – Genomes and TOPMED databases. Bioinformatics analysis showed that the sites and their corresponding amino acids were both highly conserved among vertebrates. Moreover, the influences of all the mutations on protein function were predicted to be damaging by the SIFT tool. Combining clinical data, it was found that the mutation group (93.36 µmol/L) had significantly (P = 0.038) higher total bile acid (TBA) levels than the wild-type group (40.81 µmol/L).
Conclusions
To the best of our knowledge, this is the first study to employ WES technology to detect genetic loci for ICP. Our results provide new insights into the genetic basis of ICP and will benefit the final identification of the underlying mutations.
Similar content being viewed by others


Background
Intrahepatic cholestasis of pregnancy is a pregnancy-related liver disease that mainly occurs in the second and third trimesters of pregnancy and is characterized by pruritus and abnormal liver functions [1]. The symptoms and biochemical abnormalities usually rapidly disappeared after delivery. The incidence of ICP ranges from below 1% to above 15%, with obvious regional and ethnic differences and familial clustering [2]. In China, it also reaches as high as 5.2% [3]. The recurrence rate of ICP in subsequent pregnancies reaches approximately 40% − 60% [1]. ICP increases the risk for adverse pregnancy and perinatal outcomes, including spontaneous preterm birth, intrauterine distress and amniotic fluid fecal infection [4, 5]. The serum bile acid levels in patients increase the risk of adverse perinatal outcomes [6, 7]. Therefore, understanding the molecular basis of ICP disease is very important.
Obviously, ICP is a complex disease that depends on multiple interacting factors, including genetics, endocrine hormones, nutrition and the environment [8]. In recent years, whole-genome and whole-exome sequencing have proven to be powerful new approaches to identify disease-associated variants across the full minor allele frequency (MAF) spectrum in animals [9] and humans [10]. Moreover, the 1000 Genomes Project revealed that rare variants constitute the majority of polymorphic sites in human populations [11]. In particular, accumulating evidence has demonstrated that low-frequency (0.01 ≤ MAF < 0.05) and rare (MAF < 0.01) variations often have a large effect on complex disease etiologies. Increasingly abundant examples of rare variants acting collectively for relevant quantitative traits in medicine have been noted. For example, a previous study revealed that four rare mutations of the IFIH1 gene act independently on type 1 diabetes (TID) risk [12].
Since the first ABCB4 mutation in ICP in Caucasians was reported in 1999, the efforts of many researchers have been dedicated to understanding the mechanism of ICP in many different laboratories across Europe [13]. However, deciphering the genetic basis of ICP disease is still a major challenge. To date, only a handful of causative genes (such as ABCB4 and ABCB11) [14] have been identified via genealogical analysis and Sanger sequencing. In recent years, many studies have addressed the role of the ATP8B1 and TJP2 genes in ICP susceptibility and identified some possible effect loci associated with ICP [14, 15]. Identification of the association of these genes with ICP disease is helpful to provide timely diagnosis and appropriate medical intervention for ICP pregnant women to avoid adverse maternal and fetal outcomes. Therefore, it is of great importance to identify a large number of ICP susceptibility genes that remain undiscovered.
The anoctamin family contains 10 members (ANO1-10) with two major functions: Ca2+-dependent ion channels (ANO1 and ANO2) and/or Ca2+-activated lipid scramblases with nonselective ion channel activity (ANO3-4, ANO6-8) [16,17,18]. The ANO protein family is widely expressed in eukaryotes, exhibits diverse functions in cells throughout the body and is associated with several human diseases [19]. For example, ANO1 plays roles in membrane excitability in olfactory transduction [19] and affects bile secretion and formation [20]. ANO8 encodes the transmembrane protein 16H and plays a role in the transport of glucose and other sugars, bile salts and organic acids, metal ions and amine compounds and ion channel transport, according to the functional annotation of the GeneCards. Moreover, Alaish SM et al. previously reported that ANO8 was differentially expressed in intestinal tissue between AJ (mouse strain) common bile duct ligation (CBDL) and sham-operated mice [21], suggesting that ANO8 plays a role in hepatobiliary disease. Therefore, we extrapolated and hypothesized that mutations in the ANO8 gene might affect the protein expression level and thus the transport function of bile salts.
To the best of our knowledge, only a minority of studies have addressed the genetic loci for ICP disease. However, among them, there have been no papers researching ICP with whole-exome sequencing technology. Thus, the objectives of this work were to analyze genetic mutations and putative pathogenic genes associated with clinical data in a sample of 151 Han Chinese individuals with ICP using WES data. A total of 8 mutations in the ANO8 gene were identified in eight of the 151 individuals.
Methods
Samples and clinical features
Peripheral blood samples from 151 Han Chinese ICP patients were collected from the Department of Obstetrics, Jiangxi Provincial Maternal and Child Health Hospital in Nanchang, China. A total of 27 available clinical features, including the age at diagnosis; body mass index (BMI); gestational age; the concentrations of K, Na, Cl, Ca, Mg, and P; white blood cell (WBC), red blood cell (RBC), and platelet (PLT) counts; red blood cell distribution width SD (RDW-SD); alanine transaminase (ALT), aspartate transaminase (AST), total bile acids (TBA), total bilirubin (TBIL), direct bilirubin (DBIL), indirect bilirubin (IDBIL), total cholesterol (CHOL), triglyceride (TG), high-density lipoprotein (HDL), low-density lipoprotein (LDL), and uric acid (UA) levels; newborn birth weight; Apgar score and bleeding amount were recorded. The ion concentration, liver function and lipid index were determined by an AU5800 automatic biochemical analyzer (Beckman Coulter). Routine blood tests were performed using a Sysmex-xn-2000 automatic blood cell analyzer. Summary statistics for all clinical data investigated are shown in Table 1. In addition, 1029 female control individuals without ICP were recruited. Written informed consent was obtained from each participated women in this study.
Whole-exome sequencing
A total of 151 human genomic DNA samples were isolated from peripheral blood using an Axy Prep Blood Genomic DNA Mini Prep Kit (item No. 05119KC3). DNA quality and concentration were determined by a NanoDrop-1000 spectrophotometer (Thermo Fisher, USA) and gel electrophoresis, respectively. Qualified genomic DNA samples were randomly fragmented, and the size of the library fragments was mainly distributed between 150 bp and 250 bp. End repair of DNA fragments was performed, and an “A” base was added at the 3’-end of each strand. Then, adapters were used to ligate to both ends of the end-repaired/dA-tailed DNA fragments for amplification and sequencing. Amplified DNA fragments were then purified and hybridized to a BGI Exon array. The captured products were then further amplified by circularization. Each qualified captured DNA library was then loaded on BGISEQ-500 platforms. Finally, we obtained the raw sequencing data, which were stored in FASTQ format for each individual. The informatics analysis, mainly including quality control, read mapping, variant calling, filtering and annotation, was conducted by using BWA software, GATK and ANNOVAR tool, respectively.
Statistical analysis
The t-test method was performed to analyze the potential significant differences between ANO8 mutations and wild types for the available clinical features. The P value is two sided, and the result was considered significantly different at P < 0.05. Fisher’s test was conducted to test the significance of differences in frequencies between different groups. In addition, we performed logistic regression for the IPD (individual patient data) analysis to obtain the area under the ROC curve, e.g., AUC, for the association between premature birth and TBA, ALT and AST. All the above-mentioned analyses were carried out with R software.
Evolutionary conservation analysis
The evolutionary conservation analysis of sites and amino acids was performed in 17 representative vertebrate species, human, chimpanzee, gibbon, macaque, olive baboon, mouse, rat, cow, goat, sheep, pig, dog, dingo, cat, leopard, horse, and elephant, using the genomic alignments of the Ensembl Genome Browser.
Results
The WES data results
We performed whole-exome sequencing of 151 DNA samples with an average of 14003.98 Mb of raw bases. After removing low-quality reads, we obtained an average of 139,940,436 clean reads (13991.34 Mb). The clean reads of each sample had high Q20 and Q30, which showed high sequencing quality. The average GC content was 51.20%. Figure 1 shows the base percentage composition along reads and the distributions of base quality scores on clean reads of one ICP sample (ICP66). The chromosomal positions of SNPs were based on the UCSC GRCh37/hg19.

The base percentage composition along reads and distribution of base quality scores on clean reads of ICP66. The X-axis represents positions along reads. The Y-axis is the percent (a) and quality value (b)
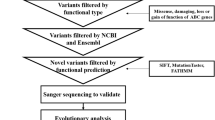
We obtained a total of 72,729 variants, including nonsynonymous, missense, splicing, start lost, stop lost/gained variants. First, we excluded variants with MAF ≥ 0.01 from the 1000 Genomes Project (http://www.internationalgenome.org/), ExAC (http://exac.broadinstitute.org/) and dbSNP ((https://www.ncbi.nlm.nih.gov/snp) databases, and 22,956 SNPs were included in subsequent analysis. In addition, 3094 variants were preserved using overlapping methods by the 1029 controls. Then, we ranked the genes and their possible damaging loci using the prediction tool SIFT to assess whether a variant affected protein function. The results implied that the gene ANO8 was prominent based on its functional annotation related to bile acid transport and pathogenicity prediction of mutations in genes, in addition to the known functional genes ABCB4, ABCB11, ATP8B1 and TJP2.
The genetic variants of ABCB4, ABCB11, ATP8B1 and TJP2
We identified a total of 61 genetic variants, including 46 intron, 6 synonymous, 8 missense, and 1 nonsense variants, in the ABCB4 gene. Among them, three variants, two missense variants, Lys386Glu and Gly527Glu, and a nonsense variant, Trp708Ter, were novel and reported for the first time. In addition, another two variants, rs1202754797 and rs201502889, were also identified in the ABCB4 gene (Table 2). For the ABCB11 gene, we observed five variants: Leu589Met, Gln605Pro, Gln1194Ter, Tyr1130Cys and Arg696Trp. The first three were newly identified mutations. After quality control, we also identified 3 and five possible pathogenic loci in ATP8B1 and TJP2, respectively. The 3 loci were Thr9Met, Gly473Arg and Arg628Trp in ATP8B1. The five variants were Arg189Ser, Ala143Thr, Arg324Trp, Arg713Trp and Gly725Glu in the TJP2 gene. For all the variants, except for Ala268Val, Ala143Thr, Arg324Trp and Gly725Glu, the remaining variants were absent in the 1000 Genomes Project and ExAC databases (Table 2).
The eight variants of the ANO8 gene
In addition, interestingly, we found that a total of eight missense mutations in the ANO8 gene in eight out of the 151 samples from patients with ICP disease (Table 3). Three of these eight mutations are known SNPs, namely, rs1316267732, rs760834212 and rs1391524054. They were identified in 30-, 23- and 24-year-old patient samples. The 30-year-old patient (ICP66) with a high TBA level (129.30 µmol/L) underwent one spontaneous abortion and had two children. The remaining five variants are novel, namely rs1, rs2, rs3, rs4 and rs5. The patients who carried rs1, rs2 and rs3 gave birth to their babies by cesarean section, while the other two patients had spontaneous abortions. Excluding the two spontaneous abortions, three out of 5 pregnant women gave birth prematurely (gestational age < 37 weeks).
In addition, 122 women of the 151 sampled women delivered their babies. Out of the 122 women, ninety individuals (73.3%, 90/122) gave birth by cesarean section, whereas 32 (26.7%, 32/122) gave birth by vaginal delivery. Thirty-two (26.7%, 32/122) delivered their babies prematurely, and 17 infants’ (13.9%, 17/122) birth weights were below 2.5 kg. Three of the 6 babies were born preterm.
Sanger sequencing to validate ANO8 variants and an additional 1029 control individuals
A total of six pairs of primers (Table 4) were designed to amplify PCR products. Then, a comparative analysis of missense mutations of the ANO8 gene was conducted by DNA sequencing from eight ICP patients and an additional 1029 control individuals with WES sequencing. Figure 2 shows the sequencing electropherograms of the known SNP rs1391524054 and the novel mutation rs1.

The sequencing electropherograms of rs1316267732 and rs1 mutations in the ANO8 gene. The mutation location is marked with an arrow
Assessing the functional impact of rareANO8 variants
These eight ANO8 mutations were absent from the 1000 Genomes Project and 1029 local controls from our hospital. Additionally, the MAFs of these mutations were low, ranging from 8e-6 to 1e-5 in three databases, e.g., ExAC, gnomAD – Genomes and TOPMED. Using Fisher’s test method, we found no significant differences in the frequencies of the variants between the 151 cases and 1029 controls (P = 0.13); in contrast, the frequencies in the databases were significantly different. This relatively lower significance between cases and controls (P = 0.13) than between cases and databases might be due to the number of samples involved.
Furthermore, we evaluated the influence of these eight mutations on protein function by using the web-available tool SIFT (http://sift.bii.a-star.edu.sg/) and obtained a score. As a SIFT score less than 0.05 is considered damaging, an amino acid substitution with such a score would be detrimental to the function of ANO8. We found that all these variants were predicted to be damaging (Table 5).
Evolutionary conservation analysis
Evolutionary conservation analysis showed that the rs1 site wild-type nucleotide allele (C) and its corresponding amino acid (proline) were both highly conserved among vertebrates, e.g., pigs, cows, sheep, dogs and cats (Fig. 3).

Evolutionary conservation analysis of the ANO8 rs1 (p. Glu325Lys) mutation. The single nucleotide C (indicating the complementary G allele) in the black solid line and the triple nucleotide CTC (indicating the GAG codon encoding proline) in the black horizontal line are highly conserved among vertebrates
Tissue expression
We used the human base website (https://hb.flatironinstitute.org) to predict ANO8 gene expression, function, regulation, and interactions in humans. The gene expression results showed that ANO8 was expressed in liver tissue with reasonable confidence (0.71). This result is consistent with the findings of the NCBI (https://www.ncbi.nlm.nih.gov/gene/57719) and GeneCards (https://www.genecards.org/cgi-bin/carddisp.pl?gene=ANO8&keywords=ANO8) websites regarding the expression of ANO8 in the liver.
To simultaneously analyze the function of ANO8, we further explored the biological process of ANO8, including the transport of inorganic anions, anions and chloride and the transmembrane transport of the above three ions. In addition, a functional network that captured liver tissue-specific interactions covering 5 data types, namely, coexpression, interaction, TF binding, GSEA microRNA targets and GSEA perturbations, from large data compendia was produced (Fig. 4). The results showed that the genes in the functional network were relevant to transport, such as EPHA1 [22], CELSR3, C10orf71, CDC14B, TM9SF4 [23], and the Wnt signaling pathway, including APC [24], IER5L, OBSL1, and MED12 [25], suggesting that the function of the ANO8 protein was likely to be related to the transport of bile salts.

The functional networks of the ANO8 gene in liver tissue. The color between genes represents the average weight of connections to the query genes
Correlations between mutations and clinical data
In the 151 ICP samples, regardless of whether the difference was significant, the mutation group tended to be associated with higher Ca2+ concentrations, platelet counts, TBA levels, TG levels, and bleeding amounts and lower birth weights (Table 6). Notably, the mutation group had significantly (P = 0.038) a higher TBA level than the wild-type group. Moreover, that of the mutation group (93.36 µmol/L) was 2-fold greater than that of the wild-type group (40.81 µmol/L).
Moreover, TBA measured by fasting peripheral blood of pregnant women is an important indicator of ICP diagnosis. The IPD analysis (Fig. 5) showed that the TBA level was more highly predictive of premature birth (AUC: 0.670 [95% CI 0548-0.768]) than the ALT and AST levels. The preterm delivery need increased at a TBA cut-off value of 46.05 µmol/L.

ROC curves for the association between premature birth and serum biochemical markers. a Association between premature birth and TBA level. b Association between premature birth and TBA, ALT, and AST levels
Discussion
So far, most reseachers make efforts to dissect the genetic architecture of ICP disease primarily focusing on ABCB4. Previously, three studies simultaneously identified ABCB4 Ile237Ile (rs2109505) as significantly associated with ICP [26,27,28]. These loci were also detected in our population. The MAFs in the 1000 Genomes Project and ExAC databases were 0.26 and 0.27, respectively. We hypothesize that this locus contributes to disease susceptibility by linkage disequilibrium between rs2109505 and the causative variant. Our study confirmed the role of ABCB11 and further expanded the role of ABCB11 gene which encoded the bile salt export pump. Our result confirmed previous studies have shown that the presence of Arg696Trp mutation in ICP population. In addition to the Arg696Trp mutation, other three novel mutations, including one prematurely stop codon Gln1194Ter, and two missense mutations Gln605Pro and Leu589Pro, were predicted as pathogenic. Besides, we did not identify any loci corresponding to ATP8B1 and TJP2 in the previous ICP literature [14, 29, 30], a reasonable explanation for this discrepancy may be the distinct genetic background and genetic heterogeneity of the populations.
Combined with clinical data, we found that 80.79% (122/151) delived the baby, in which, 26.7% (32/122) birthed prematurely and 13.9% (17/122) of the newborns weighted less than 2.5 kg. Similarily, the eight patients with ANO8 mutations having 3 newborns delivered prematurely and two spontaneous abortion The above results suggested that women with ICP had increased adverse perinatal outcome incidences, e.g., premature birth, abortion and reduced birth weight, which was consistent with the results of previous studies [2, 31]. Besides, the eight patients with ANO8 variants did not carry the possible potential effect loci of the known functional genes, ABCB4, ABCB11, ATP8B1 and TJP2, for ICP disease, implying that these ICP cases with ANO8 mutations are not caused by these mutation of functional known genes.
Bioinformatics analysis suggested that these eight variants in ANO8 gene might play an important role in the etiology of ICP disease. However, ICP disease is regulated by multiple rare variants independently or aggregatively, and further experimental verification is needed. For example, a previous study [32] employed exome array analysis to identify five new loci and low-frequency variants influencing insulin processing and secretion. Cohen et al. [33] reported that the aggregation of multiple rare variants has been associated with reduced sterol absorption and plasma low-density lipoprotein levels.
Based on the expression and function results of ANO8 combined with literature reports, the function of ANO8 was likely to be related to the transport of bile salts in the liver. Therefore, the mutations in the ANO8 gene identified in ICP women could cause bile acid transport disorder, which leads to bile acid accumulation in liver tissue. Of course, it is noteworthy that the role of the ANO8 gene and its mutations in cholestasis of pregnancy is based on bioinformatics analysis derived from WES data and network data. It remains to be determined whether ANO8 mutations cause structural and functional defects in ANO8. Therefore, subsequent cell function and in vivo experiments for ANO8 are particularly important.
Compared with wild-type group, we found that mutation group of ANO8 gene has higher TBA levels, TG levels and lower birth weights, suggesting that these mutations of the ANO8 gene might be positively involved in the pathogenesis of ICP disease. In addition, recent studies have also reported that TBA levels ≥ 40 µmol/L increased the risk of perinatal complications, such as low Apgar scores, stillbirth and preterm labor [6, 34, 35], which was consistent with our result, e.g. TBA level of 46.05 µmol/L were a critical value in increasing preterm labor.
Conclusions
In conclusion, by whole-genome sequencing analysis, we identified 18 possible pathogenic loci associated with ICP in the ABCB4, ABCB11, ATP8B1 and TJP2 genes, seven of which were novel loci. Furthermore, 8 missense mutations, including 3 known and five novel mutations, were detected in the ANO8 gene in eight of 151 Han ICP patients. To the best of our knowledge, this study is the first report revealing mutations for ICP disease by WES. By Sanger sequencing, conservation analysis, and protein functional prediction analysis, we confirmed that these variants existed and were associated with ICP. Further research should target the molecular mechanisms of these mutations in ICP disease. Our study provides new insights into the genetic architecture of ICP disease and may contribute to ICP genetic diagnosis.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ICP:
-
Intrahepatic cholestasis of pregnancy
- WES:
-
Whole-exome sequencing
- BWA:
-
Burrows-Wheeler Aligner
- GATK:
-
Genome Analysis Toolkit
- ROC:
-
Receiver operating characteristic
- MAF:
-
Minor allele frequency
- WBC:
-
White blood cell
- RBC:
-
Red blood cell
- PLT:
-
Platelet
- RDW-SD:
-
Red blood cell distribution width SD
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- TBA:
-
Total bile acid
- TBIL:
-
Total bilirubin
- DBIL:
-
Direct bilirubin
- IDBIL:
-
Indirect bilirubin
- CHOL:
-
Total cholesterol
- TG:
-
Triglyceride
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- UA:
-
Uric acid
- IPD:
-
Individual patient data
References
Ovadia C, Williamson C. Intrahepatic cholestasis of pregnancy: Recent advances. Clin Dermatol. 2016;34(3):327–34.
Ozkan S, Ceylan Y, Ozkan OV, Yildirim S. Review of a challenging clinical issue: Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2015;21(23):7134–41.
Wang XD, Yao Q, Peng B, Zhang L, Ai Y, Ying AY, Liu XH, Liu SY. [A clinical analysis of intrahepatic cholestasis of pregnancy in 1241 cases]. Zhonghua Gan Zang Bing Za Zhi. 2007;15(4):291–3.
Wikstrom Shemer E, Marschall HU, Ludvigsson JF, Stephansson O. Intrahepatic cholestasis of pregnancy and associated adverse pregnancy and fetal outcomes: a 12-year population-based cohort study. BJOG. 2013;120(6):717–23.
Yayi H, Danqing W, Shuyun L, Jicheng L. Immunologic abnormality of intrahepatic cholestasis of pregnancy. Am J Reprod Immunol. 2010;63(4):267–73.
Cui D, Zhong Y, Zhang L, Du H. Bile acid levels and risk of adverse perinatal outcomes in intrahepatic cholestasis of pregnancy: A meta-analysis. J Obstet Gynaecol Res. 2017;43(9):1411–20.
Dixon PH, Williamson C. The pathophysiology of intrahepatic cholestasis of pregnancy. Clin Res Hepatol Gastroenterol. 2016;40(2):141–53.
Williamson C, Geenes V. Intrahepatic cholestasis of pregnancy. Obstet Gynecol. 2014;124(1):120–33.
Liu X, Zhou L, Xie X, Wu Z, Xiong X, Zhang Z, Yang J, Xiao S, Zhou M, Ma J, et al. Muscle glycogen level and occurrence of acid meat in commercial hybrid pigs are regulated by two low-frequency causal variants with large effects and multiple common variants with small effects. Genet Sel Evol. 2019;51(1):46.
Panoutsopoulou K, Tachmazidou I, Zeggini E. In search of low-frequency and rare variants affecting complex traits. Hum Mol Genet. 2013;22(R1):R16–21.
Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324(5925):387–9.
Jacquemin E, Cresteil D, Manouvrier S, Boute O, Hadchouel M. Heterozygous nonsense mutation of the MDR3 gene in familial intrahepatic cholestasis of pregnancy. Lancet. 1999;353(9148):210–1.
Dixon PH, Sambrotta M, Chambers J, Taylor-Harris P, Syngelaki A, Nicolaides K, Knisely AS, Thompson RJ, Williamson C. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci Rep. 2017;7(1):11823.
Groen A, Romero MR, Kunne C, Hoosdally SJ, Dixon PH, Wooding C, Williamson C, Seppen J, Van den Oever K, Mok KS, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology. 2011;141(5):1927–37. e1921-1924.
Khelashvili G, Falzone ME, Cheng X, Lee BC, Accardi A, Weinstein H. Dynamic modulation of the lipid translocation groove generates a conductive ion channel in Ca(2+)-bound nhTMEM16. Nat Commun. 2019;10(1):4972.
Bushell SR, Pike ACW, Falzone ME, Rorsman NJG, Ta CM, Corey RA, Newport TD, Christianson JC, Scofano LF, Shintre CA, et al. The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat Commun. 2019;10(1):3956.
Reichhart N, Schoberl S, Keckeis S, Alfaar AS, Roubeix C, Cordes M, Crespo-Garcia S, Haeckel A, Kociok N, Fockler R, et al. Anoctamin-4 is a bona fide Ca(2+)-dependent non-selective cation channel. Sci Rep. 2019;9(1):2257.
Whitlock JM, Hartzell HC. Anoctamins/TMEM16 Proteins: Chloride Channels Flirting with Lipids and Extracellular Vesicles. Annu Rev Physiol. 2017;79:119–43.
Dutta AK, Khimji AK, Liu S, Karamysheva Z, Fujita A, Kresge C, Rockey DC, Feranchak AP. PKCalpha regulates TMEM16A-mediated Cl(-) secretion in human biliary cells. Am J Physiol Gastrointest Liver Physiol. 2016;310(1):G34–42.
Alaish SM, Timmons J, Smith A, Buzza MS, Murphy E, Zhao A, Sun Y, Turner DJ, Shea-Donahue T, Antalis TM, et al. Candidate Genes for Limiting Cholestatic Intestinal Injury Identified by Gene Expression Profiling. Physiol Rep. 2013; 1(4)e00073.
Yamazaki T, Masuda J, Omori T, Usui R, Akiyama H, Maru Y. EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J Cell Sci. 2009;122(Pt 2):243–55.
Perrin J, Le Coadic M, Vernay A, Dias M, Gopaldass N, Ouertatani-Sakouhi H, Cosson P. TM9 family proteins control surface targeting of glycine-rich transmembrane domains. J Cell Sci. 2015;128(13):2269–77.
Bai Y, Sha J, Kanno T. The Role of Carcinogenesis-Related Biomarkers in the Wnt Pathway and Their Effects on Epithelial-Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma. Cancers (Basel). 2020; 12(3): 555.
Zhou H, Kim S, Ishii S, Boyer TG. Mediator modulates Gli3-dependent Sonic hedgehog signaling. Mol Cell Biol. 2006;26(23):8667–82.
Dixon PH, Wadsworth CA, Chambers J, Donnelly J, Cooley S, Buckley R, Mannino R, Jarvis S, Syngelaki A, Geenes V, et al. A comprehensive analysis of common genetic variation around six candidate loci for intrahepatic cholestasis of pregnancy. Am J Gastroenterol. 2014;109(1):76–84.
Mullenbach R, Linton KJ, Wiltshire S, Weerasekera N, Chambers J, Elias E, Higgins CF, Johnston DG, McCarthy MI, Williamson C. ABCB4 gene sequence variation in women with intrahepatic cholestasis of pregnancy. J Med Genet. 2003;40(5):e70.
Wasmuth HE, Glantz A, Keppeler H, Simon E, Bartz C, Rath W, Mattsson LA, Marschall HU, Lammert F. Intrahepatic cholestasis of pregnancy: the severe form is associated with common variants of the hepatobiliary phospholipid transporter ABCB4 gene. Gut. 2007;56(2):265–70.
Giovannoni I, Callea F, Bellacchio E, Torre G, De Ville De Goyet J, Francalanci P. Genetics and Molecular Modeling of New Mutations of Familial Intrahepatic Cholestasis in a Single Italian Center. PLoS One. 2015;10(12):e0145021.
Vitale G, Gitto S, Raimondi F, Mattiaccio A, Mantovani V, Vukotic R, D’Errico A, Seri M, Russell RB, Andreone P. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J Gastroenterol. 2018;53(8):945–58.
Brouwers L, Koster MP, Page-Christiaens GC, Kemperman H, Boon J, Evers IM, Bogte A, Oudijk MA. Intrahepatic cholestasis of pregnancy: maternal and fetal outcomes associated with elevated bile acid levels. Am J Obstet Gynecol. 2015;212(1):100 e101–7.
Huyghe JR, Jackson AU, Fogarty MP, Buchkovich ML, Stancakova A, Stringham HM, Sim X, Yang L, Fuchsberger C, Cederberg H, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet. 2013;45(2):197–201.
Cohen JC, Pertsemlidis A, Fahmi S, Esmail S, Vega GL, Grundy SM, Hobbs HH. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A. 2006;103(6):1810–5.
Chen H, Zhou Y, Deng DR, Hao HY, Dang J, Li J. Intrahepatic cholestasis of pregnancy: biochemical predictors of adverse perinatal outcomes. J Huazhong Univ Sci Technolog Med Sci. 2013;33(3):412–7.
Kawakita T, Parikh LI, Ramsey PS, Huang CC, Zeymo A, Fernandez M, Smith S, Iqbal SN. Predictors of adverse neonatal outcomes in intrahepatic cholestasis of pregnancy. Am J Obstet Gynecol. 2015;213(4):570 e571–8.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Science Foundation of Jiangxi Province (No. 20192BBG70003 and No. 20171BCB24015).
Author information
Authors and Affiliations
Contributions
XL analyzed the data, prepared the figures and drafted the manuscript. HL, XZ, LN, ZL, MW and YC collected the samples. SX performed the experiments. YZ and JZ conceived and designed the experiments and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The present study followed the tenets of the Helsinki Declaration, and ethics approval was provided by the Institutional Review Board of Jiangxi Provincial Maternal and Child Health Hospital in China. Each participating woman gave written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, X., Lai, H., Zeng, X. et al. Whole-exome sequencing reveals ANO8 as a genetic risk factor for intrahepatic cholestasis of pregnancy. BMC Pregnancy Childbirth 20, 544 (2020). https://doi.org/10.1186/s12884-020-03240-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-020-03240-z