
Abstract
Background
Among the various methods available, the administration of prostaglandins is the most effective for inducing labour in women with an unfavourable cervix. Recent studies have compared treatment with various titrated doses of oral misoprostol with vaginal misoprostol or dinoprostone, indicating that the use of an escalating dose of an oral misoprostol solution is associated with a lower rate of caesarean sections and a better safety profile. The objective of this study is to assess which of these three therapeutic options (oral or vaginal misoprostol or vaginal dinoprostone) achieves the highest rate of vaginal delivery within the first 24 h of drug administration.
Methods
An open-label randomised controlled trial will be conducted in Araba University Hospital (Spain). Women at ≥41 weeks of pregnancy requiring elective induction of labour who meet the selection criteria will be randomly allocated to one of three groups: 1) vaginal dinoprostone (delivered via a controlled-release vaginal insert containing 10 mg of dinoprostone, for up to 24 h); 2) vaginal misoprostol (25 μg of vaginal misoprostol every 4 h up to a maximum of 24 h); and 3) oral misoprostol (titrated doses of 20 to 60 μg of misoprostol following a 3 h on + 1 h off regimen up to a maximum of 24 h). Both intention-to-treat analysis and per-protocol analysis will be performed.
Discussion
The proposed study seeks to gather evidence on which of these three therapeutic options achieves the highest rate of vaginal delivery with the best safety profile, to enable obstetricians to use the most effective and safe option for their patients.
Trial registration
NCT02902653 Available at: https://clinicaltrials.gov/show/NCT02902653 (7th September 2016).
Similar content being viewed by others


Background
Currently, there are pharmacological (administration of prostaglandins or isosorbide mononitrate) and mechanical (insertion of balloon catheters or cervical dilators) methods for cervical ripening [1, 2]. Prostaglandins are considered the most effective agent in women with an unfavourable cervix [3], these molecules being effective for both cervical ripening and the induction of labour [2, 4,5,6]. Their main adverse effect is excess uterine activity with or without cardiotocographic abnormalities [1, 2, 4, 6], and these effects are route of administration and dose dependent [1].
Vaginal dinoprostone
Dinoprostone, a synthetic analogue of prostaglandin E2, is available in various formulations, including intracervical gel, controlled-release vaginal systems, and vaginal tablets. The controlled-release vaginal system available in our health system is an insert containing 10 mg of dinoprostone with a release rate of 0.3 mg/h that can be left in the vagina for up to 24 h. An advantage of this system is that in the event of uterine tachysystole or cardiotocographic abnormalities, the device can be removed easily. Compared to placebo, vaginal dinoprostone is associated with a higher rate of vaginal delivery within the first 24 h of drug administration without decreasing the rate of caesarean sections, but also with a higher risk of uterine hyperstimulation with changes in foetal heart rate (FHR) [2].
Misoprostol
Misoprostol, a synthetic analogue of prostaglandin E1, is indicated for the prevention and treatment of peptic ulcer disease [7]. This drug stimulates the endometrium, induces uterine contractions and is effective for cervical ripening [1,2,3,4, 8]. In addition, its low cost and the fact that can be stored at room temperature make it a key drug for the induction of labour in developing countries [9, 10]. Misoprostol can be administered by various routes: buccal, oral, rectal, sublingual and vaginal [1, 11]. The dose and duration of administration of this drug, however, have not yet been well defined [12, 13].
Vaginal misoprostol
With vaginal administration, the concentration of misoprostol reaches a peak plasma concentration after 70 to 80 min and then decreases, levels remaining detectable up to 6 h post-administration [14]. The vaginal administration of a 50-μg dose of misoprostol has been widely studied, results indicating that this dose has a greater efficacy but is less safe than a 25-μg dose (being associated with higher rates of uterine tachysystole, uterine hyperstimulation, and caesarean section due to an abnormal cardiotocographic findings and meconium) [15, 16]. Although the World Health Organization recommends using 25 μg every 6 h, several studies have assessed the administration of this dose every 5 h, finding no increase in abnormal events [17,18,19] and a higher rate of vaginal delivery within 12 h of starting induction than with dinoprostone [11].
A 2010 Cochrane review comparing vaginal misoprostol with placebo, vaginal and intracervical dinoprostone and oxytocin showed that vaginal misoprostol at doses > 25 μg every 4 h is more effective than the other conventional methods for induction of labour, but with a higher rate of uterine hyperstimulation, while low doses of this drug (≤25 μg every 4 h) were found to have the same levels of effectiveness and risk as other methods of inducing labour [16]. In line with this, another systematic review concluded that vaginal misoprostol is more effective than vaginal dinoprostone and they have similar safety profiles [5].
Subsequently, in 2011, Silfeler et al. conducted a study comparing vaginal misoprostol (25 μg every 4 h up to a maximum of 8 doses), controlled-release vaginal dinoprostone (10 mg over 24 h) and oxytocin in women with intact membranes [18]. They observed that misoprostol was more effective than dinoprostone or oxytocin, achieving a rate of vaginal delivery of 48.5% within 12 h in the misoprostol group compared to 36.1 and 13.3% in the oxytocin and dinoprostone groups, respectively. No significant differences were found in terms of uterine hyperstimulation rate or neonatal outcomes.
More recently, in 2014, Abraham et al. carried out a retrospective study on the induction of women with premature rupture of membranes using 25 μg vaginal misoprostol every 4 h up to a maximum of 6 doses vs 10 mg of dinoprostone over 12 h [17]. They concluded that vaginal misoprostol is more effective than dinoprostone for induction of labour in this population, without increasing the rate of adverse outcomes. Further, they found a caesarean section rate of 20% in the dinoprostone group compared to 11% in the misoprostol group [17].
Oral misoprostol
Oral administration of misoprostol in solution is a route that is generally better tolerated by women since it involves fewer vaginal examinations [20]. After oral administration, misoprostol has a very short half-life of between 20 and 40 min [12]; the plasma concentration reaches an optimal level 30 min after administration and then decreases, the drug being cleared from the blood by 120 min [12, 14].
A 2011 review concluded that oral misoprostol is associated with a lower rate of caesarean sections than vaginal dinoprostone or placebo [2], and also with less uterine hyperstimulation than vaginal administration, without alterations in FHR. A 2014 Cochrane review [21] found a similar level of efficacy with oral and vaginal routes of administration but reported better perinatal outcomes with the oral route, and therefore recommended oral over vaginal administration.
Nevertheless, it is unknown what is the optimal regimen. Different studies have suggested different regimes and doses with successful but highly varied results, the rate of women achieving a vaginal birth within the 24 h of drug administration ranging from 54 to 94%. To date, it seems that titrated doses of misoprostol solution achieves better outcomes than fixed doses, as this produces constant blood levels of the drug [12].
In a clinical trial, Cheng et al. [22] compared the use of vaginal misoprostol (25 μg every 4 h) with a titrated oral misoprostol solution. The initial dose of misoprostol solution was 20 μg per hour, and this was increased by 20 μg every 4 h to a maximum of 60 μg. The rate of vaginal delivery within the first 24 h of drug administration was 94.1% with the oral route of administration, compared to 53.8% with the vaginal route, with no cases of uterine hyperstimulation. On the other hand, Rouzi et al. [12] reported a higher rate of vaginal delivery among women who received oral misoprostol (70% with misoprostol vs 55% with dinoprostone) and a lower rate of caesarean sections, but the difference did not reach significance. Finally, two recently published meta-analyses [10, 23] indicate that although vaginal misoprostol (≥50 μg) is associated with a higher success rate of vaginal delivery within the first 24 h of drug administration, a low dose of titrated oral misoprostol (< 50 μg) is associated with a lower caesarean section rate.
Since induction of labour is an increasingly widely used procedure, it is important to identify a pharmacological agent that is effective as well as safe. Given previous research findings, we expect in this open-label randomised clinical trial that the use of oral misoprostol may help improve the rate of vaginal labour while maintaining a good safety profile.
Methods/design
Aim
The objective of this study is to assess which of the three options considered, vaginal dinoprostone or vaginal or oral misoprostol, achieves a higher rate of vaginal delivery within the first 24 h after drug administration in women who undergo elective induction of labour due to their pregnancy becoming prolonged.
Participants
This single-centre open-label randomised clinical trial will be carried out in Araba University Hospital (Spain), a tertiary hospital that conducts approximately 2600 deliveries per year. The study population consists of pregnant women at 41 weeks of pregnancy or more undergoing induction of labour who meet all the inclusion criteria and none of the exclusion criteria.
-
Inclusion criteria: Women are required to be over 18 years old with a singleton pregnancy, cephalic presentation, intact membranes, a Bishop score of less than 6, and a cardiotocographic trace with a foetal heart rate pattern that is reactive with no decelerations, and provide written informed consent.
-
Exclusion criteria: To be eligible, women must not have an allergy or intolerance to any of the study drugs; a history of caesarean section or uterine surgery, or of stillbirth; high parity (four or more previous births); contraindications for vaginal birth, including placenta or vasa praevia; intrauterine growth restriction; gestational hypertension; suspected chorioamnionitis; or blood clotting disorders, epilepsy, liver or kidney disease, or moderate-to-severe heart disease.
Procedure
On the day of the induction attempt, having confirmed that women meet the selection criteria, they will be randomly allocated to one of three groups:
-
1)
Dinoprostone group: women assigned to this group will have a controlled-release non-biodegradable polymer vaginal insert containing 10 mg of dinoprostone (Propess® FERRING Laboratories-Switzerland) inserted, and the insert will be left in the patient’s vagina for a maximum of 24 h.
-
2)
Vaginal misoprostol group (Misoprostolv): women in this group will receive 25 μg of misoprostol (Misofar® BIAL laboratories, Spain) vaginally every 4 h for a maximum of 24 h (i.e., up to a maximum dose of 150 μg).
-
3)
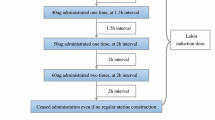
Oral misoprostol group (Misoprostolo): women in this group will be treated using a dose escalation design described in detail in Fig. 1 of this protocol. In brief, they will follow what we call a 3 + 1 regimen, corresponding to a 3-h period on the drug followed by 1 h off the drug (3 doses, one per hour + 1 h of rest).
Fig. 1 
Schedule of enrollment, interventions, and assessments of the study

At first, women will be given 20 μg of misoprostol orally every hour until adequate uterine activity is achieved (3 contractions every 10 min). If after 3 h of treatment (a total of 3 doses), uterine activity is not adequate, the drug will not be administered in the following hour, and after that 1 h of rest, one 30-μg dose will be administered each hour until adequate uterine activity is achieved, up to a maximum of three doses. If at this stage, there is still an inadequate uterine response, the same procedure will be followed, first with 40-μg doses and then with 50-μg doses, always allowing a 1-h rest period before increasing the dose. After another 1 h off, one last dose of 60 μg will be given, attempting to achieve adequate uterine activity. Once this 60-μg dose has been given, the treatment will be stopped, even if adequate uterine activity has not been achieved, and hence, in cases of inadequate uterine response, the maximum duration of this treatment would be 18 h.
In the event that women achieve adequate uterine activity at any point during the procedure, the latest dose will continue to be administered following the 3 + 1 pattern, that is, for every 3 h on the treatment, there will be 1 h off, up to a treatment duration of 24 h. Women assigned to this group will receive up to a maximum of 480 μg of oral misoprostol.
To summarise, the maximum duration of the intervention in the dinoprostone and vaginal misoprostol arms will be 24 h. In the oral misoprostol group, the intervention will also last for 24 h provided that the maximum dose is not reached, but will be stopped earlier if this dose is reached. Tables 1 and 2 summarise the treatment as a function of the progress of the induction process. Figure 1 includes some examples of the treatment. Additional files 1 and 2 include SPIRIT figure and checklist.
Additionally, in all of the groups, in the event that the maximum treatment period is completed without adequate uterine response, women will start to be given oxytocin for a maximum period of 24 h. If delivery is achieved by the end of this period, the induction will be considered a success. If induction is unsuccessful, a caesarean section will be performed.
Oxytocin infusion protocol: First, 10 units of oxytocin will be added to 500 ml of saline. The rate of infusion will be set at 3 ml/hour, doubling the dose every 20 min up to 24 ml/hour (yielding doses of 3, 6, 12 and 24 ml/h). Subsequently, the dose will be increased by 6 ml/hour every 20 min up to 120 ml/h. If uterine activity is still inadequate with this dose, 20 units of oxytocin in 500 ml of saline will be used with a starting rate of 60 ml/hour increasing the dose by 3 ml/hour every 20 min up to a dose of 90 ml/hour.
The women and their infants will be followed-up until discharge from hospital.
Randomisation
Selected women will be randomly assigned in a 1:1:1 ratio to the dinoprostone, oral misoprostol or vaginal misoprostol groups using a computer-generated random number sequence. Researchers will be blinded to this randomisation sequence throughout the study. The randomisation will be performed through an online platform that is available 24-h a day and meets data protection requirements.
Blinding
Given the characteristics of the intervention, we will not attempt to blind patients or clinicians.
Sample size calculation
Taking data in the scientific literature on the rate of vaginal delivery within 24 h after the administration of the study drugs (70% for oral misoprostol [12], 55% for dinoprostone [12] and 54% of vaginal misoprostol [24]) as a reference, we will need to analyse 333 women (111 per arm) to detect significant differences between the groups with a power of 90% and a confidence level of 95%. Assuming a 10% rate of losses during the follow-up period, we will therefore need to recruit a total of 372 women (124 per arm).
Variables
The data collected will be recorded on case report forms, safeguarding patient confidentiality at all times. The primary outcome will be the rate of vaginal delivery (normal or instrumental) within the first 24 h after drug administration.
The secondary outcomes include cervical status (Bishop score of 6 or over, assessed 12 and 24 h after drug administration), percentage of caesarean sections, use of oxytocin and, if so, the total dose, duration of latent and active phases and the pushing stage of labour, induction failure (no progress into the active phase of labour after administering oxytocin for 12 ± 3 h after membrane rupture), uterine tachysystole, hypertonicity, hyperstimulation, or rupture, adverse events including maternal and/or foetal death, pain (assessed on a visual analogue scale), women’s satisfaction with the induction process (assessed using a dichotomous question, asked 2 days after childbirth or on hospital discharge), maternal morbidity (uterine rupture, severe postpartum haemorrhage or intensive care unit admission) and foetal morbidity (1- and 5-min APGAR scores, umbilical cord pH, and neonatal asphyxia, among other variables).
Due to the extensive information about misoprostol and dinoprostone in the literature, it was considered that a data monitoring committee was not needed. We report to the ethics committees yearly on the progress of the trial. Currently, we have no plans to make any major modifications to the protocol, but we will communicate any such modifications to all people involved.
Statistical analysis
First, we will describe the general characteristics of the sample. For this, percentages with 95% confidence intervals will be calculated for qualitative data and means and standard deviations for quantitative data.
Second, logistic regression analysis will be used to assess the relationship between the intervention received and the primary outcome (vaginal delivery within the first 24 h after administration of the corresponding drug). Crude and adjusted models will be constructed using variables found to be confounding factors in the univariate analysis.
Logistic regression analysis will also be used to analyse the rates of vaginal deliveries at 12 and 48 h after starting induction and the ripening of the cervix at 12 and 24 h. The rates of maternal adverse events (gastrointestinal adverse effects, uterine tachysystole, uterine rupture, uterine hyperstimulation, morbidity, postpartum haemorrhage, and fever) and of caesarean sections will be compared between the groups with a chi-square test.
Third, multiple linear regression analysis will be performed to explore the relationships between duration of the labour (total, and of active and latent phases), the maximum level of pain reported by the woman, 1- and 5-min APGAR scores, umbilical cord pH, and the intervention (vaginal dinoprostone, and oral or vaginal misoprostol). As well as crude models, adjusted models will be built controlling for intervention group. Complications and adverse events including neonatal asphyxia, morbidity, neonatal intensive care unit admission, and mortality and other complications will be compared between groups with a chi-square test.
Both intention-to-treat and per-protocol analysis will be performed. For the intention-to-treat analysis, we will consider all women included in the study who received at least one dose of any medicine. To handle any missing data on satisfaction, we will use multiple imputation techniques, taking into account age, parity and other potentially relevant variables. We will use the IBM SPSS Statistics for Windows, version 22.0, considering a p value of < 0.05 to be significant.
Ethical considerations
Before inclusion, women will be asked to give written informed consent by their gynaecologist. This study has been approved by the Clinical Research Ethics Committee of Araba University Hospital and the Spanish Agency of Medicines and Medical Devices.
Discussion
Given that a growing percentage of women undergo induction of labour [4, 5], it is important to determine the best method for this process. There are reasons to assert that misoprostol is the best drug, since as well as being inexpensive, it can be stored at room temperature, and has high efficacy and a good safety profile, especially when administered orally [25, 26].
On the one hand, this study is one of the few that attempts to use titrated doses according to uterine response, which may result in a lower rate of hyperstimulation and caesarean sections compared to vaginal administration of misoprostol [25]. Further, despite the safety associated with the rapid clearance of oral misoprostol [27], in our trial, we will progressively increase the doses (by 10 μg) with 1 h of observation off the drug for every 3 h in which doses are given, thereby further increasing the levels of safety.
On the other hand, our formulation allows more accurate dosing than those used in other studies [12, 25, 28] In previous studies, the drug in solution has been obtained by dissolving a 200-μg tablet of misoprostol in 200 ml of water, and the solution has been kept for up to 24 h. A syringe has been used to obtain the volumes to be given the women from this solution, assuming that in 1 ml of solution there will be 1 μg of drug [12, 28] This approach does not, however, take into account the potential precipitation of the drug or that it may not be homogenously dissolved, and hence, may lead to dosing errors. In contrast, in our study, we will use 20 and 30 μg capsules, which will be dissolved in water just before administration.
If the hypothesis of this study is confirmed, oral misoprostol in titrated doses may come to be considered the most effective and safe option for pre-induction/induction of labour in women with unfavourable cervix.
Abbreviations
- CRF:
-
Case report form
- FHR:
-
Foetal heart rate
References
Sociedad Española de Ginecología y Obstetricia. Protocolo SEGO. de Induccion del parto (updated July 2013). Prog Obstet Ginecol. 2015;58:54–64.
Mozurkewich EL, Chilimigras JL, Berman DR, Perni UC, Romero VC, King VJ, et al. Methods of induction of labour: a systematic review. BMC Pregnancy Childbirth. 2011;11:84.
Blickstein I. Induction of labour. J Matern Fetal Neonatal Med. 2009;22(Suppl 2):31–7.
Alfirevic Z, Keeney E, Dowswell T, Welton NJ, Dias S, Jones LV, et al. Labour induction with prostaglandins: a systematic review and network meta-analysis. BMJ. 2015;350:h217.
Austin SC, Sanchez-Ramos L, Adair D. Labor induction with intravaginal misoprostol compared with the dinoprostone vaginal insert: a systematic review and metaanalysis. Am J Obst Gynaecol. 2010;202:624.
Stephenson ML, Hawkins JS, Powers BL, Wing DA. Misoprostol vaginal insert for induction of labor: a delivery system with accurate dosing and rapid discontinuation. Women's Health (Lond Engl). 2014;10:29–36.
Silverstein FE. Improving the gastrointestinal safety of NSAIDs: the development of misoprostol- from hypothesis to clinical practice. Dig Dis Sci. 1998;43:447–58.
Wing DA. Misoprostol vaginal insert compared with dinoprostone vaginal insert. Obstet Gynecol. 2008;112:801–12.
Ezechukwu PC, Ugwu EO, Obi SN, Chigbu CO. Oral versus vaginal misoprostol for induction of labor in Enugu, Nigeria: a randomized controlled trial. Arch Gynecol Obstet. 2015;291(3):537–44.
Chen W, Xue J, Peprah MK, Wen SW, Walker M, Gao Y, Tang Y. A systematic review and network meta-analysis comparing the use of Foley catheters, misoprostol, and dinoprostone for cervical ripening in the induction of labour. BJOG. 2016;123(3):346–54.
Özkan S, Calişkan E, Doğer E, Yücesoy I, Ozeren S, Vural B. Comparative efficacy and safety on vaginal misoprostol versus dinoprostone vaginal insert in labor induction at term: a randomized trial. Arch Gynecol Obstet. 2009;280:19–24.
Rouzi AA, Alsibiani S, Mansouri N, Alsinani N, Darhouse K. Randomized clinical trial between hourly titrated oral misoprostol and vaginal dinoprostone for induction of labor. Am J Obstet Gynecol. 2014;210:e56.e1–6.
Kundodyiwa TW, Alfirevic Z, Weeks AD. Low-dose Oral misoprostol for induction of labor: a systematic review. Obstet Gynecol. 2009;113(2 Pt 1):374–83.
Tang OS, Gemzell-Danielsson K, Ho PC. Misoprostol: pharmacokinetic profiles, effects on the uterus and side-effects. Int J Gynecol Obstet. 2007;99:S160-7.
McMaster K, Sanchez-Ramos L, Kaunitz AM. Balancing the efficacy and safety of misoprostol: a meta-analysis comparing 25 versus 50 micrograms of intravaginal misoprostol for the induction of labour. BJOG. 2015;122(4):468–76.
Hofmeyr GJ, Gülmezoglu AM, Pileggi C. Vaginal misoprostol for cervical ripening and induction of labour. Cochrane Database Syst Rev. 2010;10:CD000941.
Abraham C, Meirowitz N, Kohn N. Labor induction for premature rupture of membranes using vaginal misoprostol versus Dinoprostone vaginal insert. Am J Perinatol. 2014;31:181–6.
Silfeler DB, Tandogan B, Ayvaci H, Silfeler I, Yenidede I, Dayicioglu V. A comparison of misoprostol, controlled-release dinoprostone vaginal insert and oxytocin for cervical ripening. Arch Gynecol Obstet. 2011;284:1331–7.
Haghighi L, Homan H, Raoofi Z, Najmi Z. Intravaginal isosorbide dinitrate or misoprostol for cervical ripening prior to induction of labour: a randomised controlled trial. J Obstet Gynaecol. 2013;33:272–6.
Aceituno Velasco L, Sanchez Barroso MT, Segura García MH, Gonzalez Acosta V, De la Fuente Pedrosa R, Barqueros Ramirez A, et al. Efficacy and safety of oral solution dosed misoprostol versus misoprostol vaginally in labour induction. Open Journal of Obstetrics and Gynecology. 2013;3:673–9.
Alfirevic Z, Aflaifel N, Weeks A. Oral misoprostol for induction of labour. Cochrane Database Syst Rev. 2014;6:CD001338.
Cheng SY, Ming H, Lee JC. Titrated oral compared with vaginal misoprostol for labor induction. A randomized controlled trial Obstet Gynecol. 2008;111:119–25.
Alfirevic Z, Keeney E, Dowswell T, Welton NJ, Medley N, Dias S, Jones LV, Caldwell DM. Methods to induce labour: a systematic review, network meta-analysis and cost-effectiveness analysis. BJOG. 2016;123:1462–70.
Murchison A, Duff P. Misoprostol for uterine evacuation in patients with early pregnancy failures. Am J Obstet Gynecol. 2004;190:1445–6.
Bricker L, Peden H, Tomlinson AJ, Al-Hussaini TK, Idama T, Candelier C, et al. Titrated low-dose vaginal and/or oral misoprostol to induce labour for prelabour membrane rupture: a randomised trial. BJOG. 2008;115:1503–11.
Alfirevic Z, Keeney E, Dowswell T, Welton NJ, Medley N, Dias S. Which method is best for the induction of labour? A systematic review, network meta-analysis and cost-effectiveness analysis. Health Technol Assess. 2016;20:1–584.
Tang OS, Ho PC. The pharmacokinetics and different regimens of misoprostol in early first-trimester medical abortion. Contraception. 2006;74:26–30.
Wang X, Yang A, Ma Q, Li X, Qin L, He T. Comparative study of titrated oral misoprostol solution and vaginal dinoprostone for labor induction at term pregnancy. Arch Gynecol Obstet. 2016;294:495–503.
Acknowledgements
We are grateful to study participants, providers research nurses, and research assistants for their contributions. We would like to acknowledge Ines Perez Francisco for her support in this study.
Funding
No external funding has been received for this research.
Availability of data and materials
Regarding sharing materials and managing of intellectual property, data will be available on request to the authors. Data sharing is not applicable to this article as no datasets have yet been generated or analysed.
Author information
Authors and Affiliations
Contributions
OLO, LU ALP and IL conceived and designed the study. FSR, OE, JAS, AF, IB, LB, RHC, DPC, JALL and JV critically evaluated the manuscript. All authors significantly contributed to the development and implementation of the protocol. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Women will be asked to sign the corresponding informed consent form by their gynaecologist before inclusion. This study has been approved by the Clinical Research Ethics Committee of Araba University Hospital and the Spanish Agency of Medicines and Medical Devices.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
SPIRIT figure. (PPTX 332 kb)
Additional file 2:
SPIRIT checklist. (DOC 123 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lapuente-Ocamica, O., Ugarte, L., Lopez-Picado, A. et al. Efficacy and safety of administering oral misoprostol by titration compared to vaginal misoprostol and dinoprostone for cervical ripening and induction of labour: study protocol for a randomised clinical trial. BMC Pregnancy Childbirth 19, 14 (2019). https://doi.org/10.1186/s12884-018-2132-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-018-2132-3