
Abstract
Background
Persistent post-concussion symptoms (PPCS) affect between 34 and 46% after a mild traumatic brain injury (mTBI). Many also experience exercise intolerance. Sub-symptom threshold aerobic exercise, SSTAE (exercise at an intensity level that does not increase symptoms) is proposed as a treatment to both reduce the symptom burden and increase the exercise tolerance after the injury. It is unclear if this also applies in a more chronic phase after mTBI.
Main purpose
The main purpose of this study is to evaluate whether SSTAE in addition to ordinary rehabilitation will lead to clinically meaningful improvement of symptom burden, normalize exercise tolerance, increase physical activity, improve health-related quality of life, and reduce patient-specific activity limitations compared to a control group that only receives ordinary rehabilitation.
Design
Randomized, controlled, single-blind parallel-group study with three measurement times; T0 at baseline, T1 after the intervention and T2 six months after T1.
Methods
Patients between the ages of 18 and 60 with exercise intolerance and persistent PPCS (> 3 months) will be recruited to the study and randomized to two groups. All patients will receive follow-up at the outpatient TBI clinic. The intervention group will in addition receive SSTAE for 12 weeks with exercise diaries and a retest every 3 weeks for optimal dosage and progression. The Rivermead post-concussion symptoms questionnaire will be the main outcome measure. The secondary outcome measure will be a test of exercise tolerance—the Buffalo Concussion Treadmill Test. Other outcome measures include the patient-specific functional scale that measures patient-specific activity limitations, as well as outcome measures for diagnosis-specific health-related quality of life, anxiety and depression, specific symptoms such as dizziness, headache and fatigue, and physical activity.
Discussion
This study will add knowledge about the effect of SSTAE and whether it should be implemented in rehabilitation for the adult population with persistent PPCS after mTBI. The nested feasibility trial showed that the SSTAE intervention was safe and that the study procedures and delivery of the intervention overall were feasible. However, minor amendments to the study protocol were made prior to the commencement of the RCT.
Trial registration
Clinical Trials.gov, NCT05086419. Registered on September 5th, 2021.
Similar content being viewed by others


Background
Exercise, as a rehabilitation intervention for patients with persistent symptoms after mild traumatic brain injury (mTBI), will be explored in this randomized controlled trial (RCT). Traumatic brain injury (TBI) is a complex injury defined as a change in brain function or other signs of brain pathology caused by an external force [1]. Between 80–90% are mTBI, with an annual incidence in Norway of 302 pr. 100,000 [2].
The most common causes of mTBI injuries are falls, traffic, sports related accidents and violence [3,4,5]. Many recover well during the first three months after the injury, but between 34 and 46% report cognitive, psychological, and physical persistent post-concussion symptoms (PPCS) at three and six months [6]. More specifically, headache, dizziness, fatigue, emotional distress, problems with memory, concentration and slowed thinking are common PPCS [6, 7]. In addition, exercise intolerance, PPCS exacerbation during physical activity, is challenging to manage. Because many patients with mTBI are young and of working age [2], it is important to develop effective rehabilitation interventions.
Traditionally, mTBI management has consisted of advice on physical and cognitive rest until the PPCS wanes, and then gradually resume activities [7,8,9]. However, this approach is questioned [7] as studies show no effect of rest on PPCS after mTBI [10,11,12]. Studies rather suggest that cognitive and physical rest beyond the first few days contributes to persistent PPCS [8, 11]. PPCS might also reduce activity and participation, and is associated with reduced health-related quality of life [13]. Hence, a more physically active approach in rehabilitation after mTBI should be explored [8, 9, 14].
Studies indicate that both central and systemic physiological dysfunction can contribute to exercise intolerance after mTBI [9, 14]. Exercise intolerance can be understood as reduced ability to be physically active/exercise at the expected level according to age and physical condition due to exacerbation of symptoms [14,15,16]. Research suggests that metabolic and physiological changes in the acute phase after mTBI may lead to autonomic dysfunction and abnormal control of cerebral blood flow. This can cause increased symptoms (e.g., headache and dizziness) with exertion and lead to exercise intolerance [9, 14]. The risk of developing exercise intolerance with symptom exacerbation during physical activities is believed to increase 3–18 times after mTBI compared to healthy controls [14, 15]. However, it is unclear whether this also applies in a more persistent phase after mTBI.
Aerobic exercise at an intensity level that does not increase symptoms (sub-symptom threshold aerobic exercise, SSTAE) has received increasing focus in rehabilitation of patients with PPCS after mTBI. Recent studies suggest that SSTAE is safe, reduces PPCS, and may contribute to fewer activity limitations and increased participation [7, 17, 18]. However, studies utilizing an adapted exercise program have mainly studied young athletes [7, 19] or general instructions about physical activity to the mTBI adult population in the acute phase as mean to prevent PPCS [20]. The Buffalo Concussion Treadmill Test (BCTT) is a safe and validated treadmill test that has been developed to evaluate exercise intolerance after mTBI [8, 9, 21, 22]. The BCTT is designed to identify symptom exacerbation thresholds while SSTAE may be utilized to guide the exercise intensity based on the symptom threshold, i.e., sub-symptom exercising [8, 9, 14, 15, 17, 21,22,23].
To our knowledge, no larger clinical trials determining the effects of SSTAE in patients suffering from exercise intolerance and PPCS after mTBI in the general adult population have currently been concluded [7, 24]. Hence, there is a lack of evidence-based knowledge about the effect of SSTAE in adult patients in a more chronic phase after the injury. Few larger studies with control groups have made it difficult to draw good evidence-based guidelines for clinical practice [7]. Thus, there is a need for randomised clinical trials (RCT) testing the effect of SSTAE in the adult population with PPCS. The main purpose of this study is to explore the effect of SSTAE on PPCS and exercise intolerance in patients with mTBI compared with a control group receiving treatment as usual (TAU) only.
Objectives
The aim of this study is to explore whether SSTAE, in addition to TAU, will reduce symptom burden and normalize exercise tolerance compared with a control group that only receives TAU.
Further, we aim to evaluate the effect of SSTAE on changes in symptoms such as headache, fatigue, anxiety, and depression, and to register changes in physical activity, health-related quality of life and patient-specific activity limitations.
Our specific hypotheses are:
-
H1: The SSTAE intervention will result in a greater reduction in PPCS symptom burden compared with TAU only.
-
H2: The SSTAE intervention will result in a greater normalisation of exercise tolerance compared with TAU only.
-
H3: The SSTAE intervention will result in greater improvement in headaches, fatigue, anxiety, depression, physical activity, and health-related quality of life compared with TAU only.
-
H4: The SSTAE intervention will result in greater improvement in patient-specific activity limitations compared with TAU only.
In accordance with the New Medical Research Council guidelines [25] we conducted a feasibility study as a preparatory step before initiating the RCT. The feasibility study is nested in this protocol article. The aim of the feasibility study was to evaluate the intervention arm using predefined success criteria shown in Table 2.
Trial design
This is as a randomized, controlled, single-blind clinical trial with two parallel groups allocated with a 1:1 ratio, and a primary endpoint of difference in PPCS burden at 12 weeks. See Table 1 for Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) [26, 27].
Patients referred to the TBI outpatient clinic will be screened for eligibility by the treating physician and invited to participate in the study. Informed consent will be collected and baseline assessment (T0), including patient reported outcome measures and test of exercise tolerance, will be conducted (Table 3). After the baseline assessment, participants will be randomized to SSTAE intervention or TAU group. Further, follow-up testing will be carried out after the intervention period at 12 weeks (T1) and 6 months (T2) after the baseline assessment.
The nested feasibility study was a non-randomized, feasibility study of the intervention arm of the RCT, and included baseline assessment and follow-up after 12 weeks of SSTAE. According to the predefined success criteria, we assessed the recruitment process, retention rates, outcome measures, and adherence to the intervention. A convenience sample including 11 patients with mTBI and self- reported exercise intolerance was recruited to the feasibility study from October – November 2021.
Methods: patients, interventions, and outcomes
Study setting
The clinical trial will be carried out at the TBI outpatient clinic at the Department of Physical Medicine and Rehabilitation, Oslo University Hospital (OUH), the trauma referral centre in South-East Norway [28]. Patients are referred to the TBI outpatient clinic from the Emergency department, Neurosurgical department, and from general practitioners. Assessments and clinical testing will be performed in an exercise laboratory at the TBI outpatient clinic. The exercise program will be conducted in the patients’ home setting. The feasibility study was carried out within the same study setting.
Eligibility criteria
The following inclusion and exclusion criteria will be applied in this trial:
-
Inclusion criteria: Patients 18–60 years diagnosed with mTBI as defined by the World Health Organization Collaborating Center Task Force [29] with persistent PPCS 3 – 24 months post injury and reduced tolerance of physical activity, defined as self-reported exacerbation of PPCS during physical activity and exercise.
-
Exclusion criteria: Patients with neurological or severe psychiatric conditions documented in the medical record, cardiovascular disease that contraindicates exercise testing [30], extremity injuries that prevent physical exercise, ongoing substance abuse, and insufficient understanding of Norwegian to follow instructions or fill in forms will be excluded before baseline testing. Additionally, patients with no self-reported exacerbation of PPCS during the BCTT will be excluded.
-
In the feasibility study, patients who did not fulfil the stop criteria for the BCTT but experienced symptom exacerbation within 24 h after the test were also included.
Consent
All patients will receive written and oral information about the study from the treating physician or physiotherapist prior to the baseline assessment and provide their written informed consent. The procedure for obtaining informed consent in the feasibility study was consistent with the current protocol.
Interventions
The Buffalo Concussion Treadmill Test—BCTT
The BCTT is the secondary outcome in this trial, but it is also an essential part of the SSTAE intervention and will therefore be described here. The BCTT is a standardized incremental treadmill test designed to identify exercise intolerance and establish symptom thresholds in mTBI patients [17, 21, 22, 31]. The patient walks at a brisk pace on a treadmill (5.2 – 5.8 kph), as incline is increased by 1% each minute. If 15% incline is reached, the speed is increased by 0.3 km/h each minute. The PPCS rated on a 0–10 (best–worst) numerical rating scale (NRS), perceived exertion on Borg Ratings of Perceived Exertion (RPE) 6–20 (lowest to highest) [32], and heart rate (HR) are recorded at the test’s baseline and each minute. The BCTT is terminated when one of three criteria is reached: 1) ≥ 3 points increase in PPCS on the NRS; 2) Borg RPE ≥ 18; 3) 90% of age estimated max HR calculated by (211–0.65 × age) [33]. The first criterium is interpreted as exercise intolerance, and the associated HR is the symptom threshold. The test is considered negative for exercise intolerance if terminated by the intensity criteria, and these patients will be excluded. In the feasibility study, the BCTT was administrated with stop criteria 1 and 2. The third stop criterion was added for the planned RCT to ensure that the participants did not exceed 90% of their estimated HRmax level. We also revised the symptom registration from registration of individual symptoms to registration of an overall PPCS score on the NRS to ensure a total symptom exacerbation of NRS ≥ 3 points.
Sub-symptom threshold aerobic exercises (SSTAE)
The SSTAE intervention comprises consultations, BCTT testing and a personalized exercise program. The patients are instructed to perform aerobic exercise in their preferred mode, at an intensity of 80–90% of the symptom threshold, as identified during the BCTT, i.e., at the sub-symptom threshold [22, 23]. Each session follows this plan: 5–10 min warm up, 20 min aerobic exercise at sub-symptom threshold intensity, followed by 5–10 min of active cool down. The intensity of the SSTAE is monitored using a HR monitor and/or the Borg RPE scale. If symptoms exacerbate during an exercise session with ≥ 3 NRS, or perpetuate the following days, the patients are advised to reduce the HR with 5–10 beats/minute to reduce the PPCS. Exercise frequency is jointly decided by patient and therapist, from 3 to 5 sessions per week, based on exercise intensity, symptom burden and other external loads. The patients are encouraged to contact physiotherapist (LJVV) by phone for advice and reassurance if necessary. If needed, patients will be referred to a physiotherapist in primary health care service for exercise adherence.
To ensure appropriate exercise dosing, adherence and progression, the patients are offered two guided treadmill sessions during the first two weeks of the intervention in addition to a new BCTT at week 3, 6, 9 prior to T1 after 12 weeks. Compliance with the SSTAE will be recorded using an exercise diary and constitute the basis for the consultations focusing on supporting, motivating, and reassuring the patients. The control group is followed up with assessments and BCTT at baseline (T0), after 12 weeks (T1) and six months (T2) only.
In the feasibility study, the described SSTAE intervention arm was implemented and tested on all included patients.
Treatment as usual
All patients included in the study will receive TAU at the TBI outpatient clinic. The TAU comprises individual contacts and an educational group provided by a multidisciplinary team. The specific treatment each participant receives varies according to individual needs. A specialist in physical medicine and rehabilitation addresses physical problems related to the injury, while a neuropsychologist addresses psychological or cognitive complaints. An occupational therapist helps the patients structure their day and a social worker advise patients on issues relating to work, legal rights, and benefits. A physical therapist addresses vestibular symptoms and physical activity. In addition, the educational group entails meeting 2 h once a week over a period of 4 weeks and addresses general information about mild-to-moderate TBI, common symptoms and problems in daily life, and advice regarding how to manage these [34]. The TAU includes general advice on physical activity based on recommendations from the Norwegian Directorate of Health, but not specific guidance on SSTAE and help with exercise dose.
Feasibility of the recruitment process and the intervention arm of the RCT
The nested feasibility study in this protocol article was based on pre-defined objectives with success criteria grounded on recommendations from literature [25, 35] and experience from previous clinical trials at the department of Physical Medicine and Rehabilitation. The procedures described in the protocol for T0 at baseline to T1 at 12 weeks were followed.
Eighteen patients were consecutively assessed for eligibility, five declined participation, leaving 13 for the baseline assessment. Two were excluded due to negative BCTT or aberrant HR during BCTT, leaving 11 patients, eight women and three men, with exercise intolerance included in the feasibility study. Two patients were lost to follow up during the intervention.
The mean age of the 11 patients completing the baseline assessment with exercise intolerance was 36.9 (SD 11.4) years. All had Glasgow Coma Scale score of 15 at the time of injury; one of them had intracranial injury on CT scan. Mean time since injury was 5.1 (SD 2.5) months. Five patients sustained their injuries from falls, two by sports-related activities, one from violence and three from miscellaneous situations. The predefined success criteria and the results from the feasibility study are presented in Table 2.
The results for the primary and secondary outcomes in the feasibility study are presented in more detail in the Supplementary file.
Outcomes
The primary outcome is symptom burden measured with the Rivermead Post-Concussion Symptoms Questionnaire (RPQ). The secondary outcome is exercise intolerance measured with the BCTT. Other outcomes include headache symptoms and severity, fatigue, anxiety, psychological distress, depressive symptoms, estimates of physical activity, health-related quality of life and activity limitations, and adherence to the intervention. The selected outcome measures have satisfactory psychometric properties. The outcomes with references are displayed in Table 3. The outcomes will be administered at baseline (prior to group allocation), at 12 weeks and 6 months after baseline and the order of administration will be standardized. Adherence to the intervention will only be measured in the intervention group. In the feasibility study the same Patient Reported Outcome Measures (PROMs) were administrated with a few changes in the RCT (see Table 3).
Primary outcome
The RPQ [36] is a 16 items questionnaire about post-concussion symptoms rated on a 5-point scale from 0–4 where 0 = not experienced to 4 = severe problem. The scale range is 0–64 points (best–worst). The items cover physical symptoms (headache, dizziness, nausea, noise sensitivity, sleep disturbance, vision, and light sensitivity) and emotional/cognitive symptoms (fatigue, psychological distress, irritability, frustration, memory, concentration, speed of thinking, restlessness). The primary endpoint is the difference in change of symptom burden on the RPQ between the SSTAE and control groups from baseline to 12 weeks, with a follow up at 6 months after baseline.
Secondary outcome
The BCTT is described in detail above under the intervention. Changes in BCTT as reached percentage of estimated HRmax and changes in duration of the test from baseline (T0) to 12 weeks (T1), is the secondary endpoint.
In addition to outcome measures displayed in Table 3, demographic (age, sex, level of education, marital status, occupation, and sick leave status) and injury related variables (GCS, imaging finding, loss of consciousness (LOC), post-traumatic amnesia (PTA) and cause of injury) will be registered. Treatments outside of TAU initiated by the hospital or the patients themselves during the study period will be registered.
Participant timeline
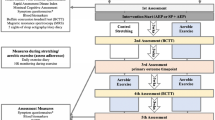
A study flowchart is provided in Fig. 1.

Flowchart of the RCT.
PROMS: Patient Reported Outcome Measures; BCTT: Buffalo Concussion Treadmill Tes; SSTAE: Sub symptom threshold aerobic exercise; TAU: Treatment as usual
Sample size
Based on RPQ and results from a pilot study at OUH (53), a power calculation was conducted with a SD of 8.7 and a mean group difference of 5.0 points on the RPQ. Given an 80% power and an alpha of 0.05, 96 participants should be included to find significant improvement. With an anticipated dropout rate of 20%, we will recruit 120 participants for this trial (60 in each group). No interim analysis is planned to change sample size during the recruitment period.
Recruitment
Potential participants will be recruited from the TBI outpatient clinic at OUH. They will be informed about the study, screened for inclusion and exclusion criteria, and invited to participate by the treating physician or rehabilitation professionals. Eligibility will be confirmed at the baseline assessment prior to randomization. Approximately 270 patients between 18 – 60 years with mTBI are treated at the clinic per year. About 30% will meet the inclusion criteria, meaning approx. 80 patients pr. year will be qualified. If necessary, potential participants will be recruited from the TBI outpatient clinic at Sunnaas Rehabilitation Hospital to reach the target sample size.
Assignment of interventions: allocation
Sequence generation
Patients will be randomized in a 1:1 ratio to the intervention or control group. To ensure allocation concealment, a computer-generated list with randomised variable block sizes will be prepared by an independent statistician. Sequentially numbered, opaque, sealed envelopes containing the allocation will be prepared by the senior researcher (HLS) and drawn consecutively after the baseline assessment.
Concealment
The allocation sequence list will be stored by a senior researcher not involved in the execution of the intervention. The therapist (LJVV) assigning the participants to the randomization, and the blinded assessors do not have access to this list.
Implementation
Eligible patients will be identified by the treating physician or rehabilitation professionals at the TBI outpatient clinic at OUH. The therapist delivering the intervention receives information about the potential eligible patients and contacts them by phone for further information and for scheduling the baseline assessment. The therapist will assign a study ID number to each participant scheduled for baseline assessment. Subsequently, when included in the study, the patients will be assigned a randomization number that is different from the study ID number. Only the senior researcher (HLS) will have access to the computer-generated list with randomization numbers.
Blinding
Blinding of the patients or therapist to the treatment allocation is not possible. However, the randomisation occurs after baseline testing (T0), and the follow-up tests at T1 and T2 will be performed by an assessor blinded to the group allocation. The patients will be instructed not to disclose their group allocation. For the statistical analyses, dummy id-numbers will be used, and an independent statistician, blinded for the group allocation, will be responsible for the primary analyses of the data.
Data collection, management, and analysis
Plans for assessment and collection of outcomes
The outcome assessors will be trained in the administration of all outcome measures. The data collection will occur at the three time points, baseline (T0), and 12 weeks (T1) and six months (T2) after baseline. The PROMs will be filled out and collected before the interview-based questionnaire and the BCTT. The PROMs will be checked for any missing items and if required, be completed by the patient.
Plans to promote participant retention and complete follow-up
To ensure adherence to the planned timing of the retests and follow-ups, one assigned researcher (LJVV) will administer all patient appointments.
To minimize control group discontent of not being allocated to the SSTAE intervention and potential loss to follow-ups, the control group will receive information about and be offered guidance in SSTAE after T2 if interested.
Data management
All data material will be assigned an ID number. Only the project researchers will have access to list linking participants names to ID numbers. The de-identified documents will be stored according to national regulations. The de-identified documents will be transferred to an electronic data file on a research server at OUH in accordance with the Norwegian Data Protection Law. Only researchers that are actively contributing to statistical analyses and publication will have access to the final data set. Data entry quality will be ensured through initial exploratory analyses.
Statistical methods
Statistical analyses will be performed in collaboration with an independent statistician at Oslo Centre for Biostatistics and Epidemiology at OUH. Updated IBM SPSS and RStudio software will be used for the statistical analysis.
Primary and secondary outcomes
Descriptive statistics will be used on socio-demographic and injury-related data. To analyze whether the intervention has a positive additional effect on symptom burden and exercise intolerance compared to the control group, a mixed-model analysis will be used. In this analysis, time (T0-T2) will be used as a «repeated-measures factor» and comparison between the intervention and the control group as a «between-group factor». The linear mixed model analyses will give estimated mean values with 95% confidence intervals for all time points (T0-T2) for each group. Estimates of mean between group changes from T0 to T1 and T2 will be provided. To establish treatment effect, the analysis of primary interest is a time x group interaction in the direction of the intervention group improving above the levels of the control group at T1. A significance level of 0.05 will be applied.
To reduce the risk of dropout bias, the analyzes will be performed on an intention-to-treat basis.
By including all participants in the analysis in the group they were randomized to.
The same mixed-model analyses will be performed for the PROMs described in our third hypothesis (H3).
Methods for any additional analyses (subgroups and adjusted analyses
The baseline population, including patients that are excluded due to a negative BCTT, will be described and analysed. Group differences between patients with and without a negative BCTT will be explored. Factors of PPCS and exercise intolerance at baseline will be analysed with multivariable regression analyses. Adherence to the intervention and changes on the BCTT will be described in the intervention group.
Methods to handle missing data
Missing data will be handled by questionnaire specific imputation, last observation carried forward and multiple imputation techniques. In the mixed-model analyses missing data will be handled by the analysis using the maximal likelihood approach under the assumption of missing at random.
Data monitoring
Composition of the data monitoring committee and its role and reporting structure
An external data monitoring committee was considered unnecessary due to the small sample size and a relatively short timeframe of the intervention and follow-ups. To ensure adherence to the protocol, the date and time of all assessments and follow-ups are documented in both the intervention and control groups.
Adverse event reporting and harms
Any harms and/or serious adverse events will be registered and reported in future publications.
Frequency and procedures for auditing trial conduct
Not applicable.
Ethics and dissemination
Research ethics approval
The study is approved by the regional ethical committee for Medical and Health Research Ethics (#256,109) and the OUH Data Protection Officer/Norwegian Center for Research Data. The project will be conducted according to the Helsinki declaration. The planned RCT and the feasibility study are registered at clinicaltrial.gov #NCT05086419.
Confidentiality
Access to the list that can re-identify the patients will be restricted to a senior researcher (HLS).
Participant information will be handled by health-care professionals adhering to Norwegian law on confidentiality.
Declaration of interests
No financial or other competing interests.
Ancillary and post-trial care
All patients are covered by the Norwegian System of Patient Injury Compensation.
Protocol amendments
Important amendments to the protocol will be approved by the regional Committee for Medical and Health Research Ethics and reported to the Data protection office at OUH. Amendments will be made to the clinicaltrials.gov registry.
Dissemination policy
Results from this project will be disseminated through presentations at national and international conferences and published articles. It is expected that the study will result in 3–5 articles published in internationally recognized peer reviewed journals in the fields of physiotherapy, exercise physiology, sports medicine, and rehabilitation. The results will also be shared with user organizations.
The Consolidated Standards of Reporting Trials (CONSORT) will be applied to facilitate transparency and critical appraisal of the trial. Authorship criteria will adhere to the recommendations from the International Committee of Medical Journal Editors (ICMJE).
The project has a plan for user participation which is in line with the OUH research strategy. The goal is to ensure that users' perspectives, needs, and experiences are reflected in the project.
Discussion
This RCT will contribute to increased knowledge about the effect of SSTAE on persistent PPCS and exercise intolerance after mTBI. If proven effective, the trial will support the implementation of SSTAE as part of the rehabilitation for this patient group. In particular, the trial will provide knowledge about the generalizability of the intervention to the adult population with persistent PPCS after mTBI, in addition to replicating the effectiveness of the SSTAE found in other studies mainly on young athletes. Furthermore, the trial will have the potential to expand our knowledge about how reduced symptom burden through SSTAE may influence quality of life, and activity limitations in this group of patients.
The nested feasibility trial showed that BCTT and the SSTAE intervention was safe and that the study procedures and delivery of the intervention overall were feasible. However, minor amendments to the study protocol were made prior to the commencement of the RCT.
Based on the feasibility study, the recruitment process was considered satisfactory for the planned RCT with a consent rate of 72% which was well above the success criteria recommending a consent rate of > 50%. Furthermore, the drop-out rate of 18% was acceptable as we consider attrition rates between 15–20% as a low risk of bias [50, 51].
Completion time on the questionnaires was acceptable as 92% of the patients answered within 40 min with less than 10% missing items. However, because some patients reported higher symptom intensity after completing the outcome measures, the QOLIBRI was replaced with the shorter version QOLIBRI-OS [45] to reduce the patient burden. Furthermore, the DHI was removed because only a few patients experienced dizziness and this questionnaire was not suitable as it assumes that the respondents have dizziness [52]. Moreover, dizziness is assessed with a question on the RPQ [36] and registered under the BCTT if present.
Because patients may experience increase in several symptoms during the BCTT, we determined to use a pooled-symptom NRS score in the RCT to simplify the scoring process. Due to cardiovascular risk [30] and risks of symptom exacerbation after BCTT we changed the BCTT stop criteria from voluntary exhaustion to 90% of estimated max HR or ≥ 18 Borg RPE. This will probably result in more patients being excluded due to a normal BCTT; however, inactive patients will not be tested towards maximum intensity, and thus, feel safer and the test less provocative [30].
Adherence to the intervention arm in the feasibility study was satisfactory with over 90% attendance rate of SSTAE and BCTT follow-ups. The prescribed home exercises were performed by 75%, and almost all maintained the exercise diary. For the full-scale RCT, we added the Problematic Experience of Therapy Scale (PETS) that will enable us to identify factors that may influence non-adherence [46].
Results from the primary and secondary outcomes were satisfactory in that they showed that most patients in the feasibility study reported reduced symptom burden and were able to exercise at a higher intensity level after 12-weeks of SSTAE. This is in line with a former study that also found that SSTAE contributed to decreased symptom burden, increased exercise tolerance, increased physical activity, and increased health-related quality of life [53] and thus represents a promising option for treatment of persistent PPCS.
A strength of the feasibility study was that the aspects of feasibility were assessed based on specific predefined criteria, which is in line with guidelines [54, 55]. However, the recruitment period was limited to two months and included a small convenience sample. Thus, the generalizability of the feasibility study might be limited. On the other hand, we tested 8% of the sample size required in the RCT which is considered sufficient data to indicate necessary amendments [56]. Furthermore, the findings should be interpreted with caution due to a lack of a control group. However, the purpose was not to determine the effect of the intervention, but to assess its feasibility.
Trial status
Recruitment for the RCT began in March 2022 and will continue until target sample size has been reached, estimated by the end of 2023.
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available due to restrictions from the regional ethical committee but could be made available upon requests through the principal investigator (Ingerid Kleffelgård, inff@live.no).
Abbreviations
- mTBI:
-
Mild Traumatic Brain Injury
- PPCS:
-
Post-Concussion Symptoms
- BCTT:
-
Buffalo Concussion Treadmill Test
- SSTAE:
-
Sub-Symptom Threshold Aerobic Exercise
- PROM:
-
Patient Reported Outcome Measures
- RPQ:
-
Rivermead Post Concussion Symptoms Questionnaire
- GCS:
-
Glasgow Coma Scale
- HR:
-
Heart Rate
- RCT:
-
Randomized controlled trial
- TAU:
-
Treatment as usual
- NRS:
-
Numerical Rating Scale
- KPH:
-
Kilometer per hour
- RPE:
-
Rating of Perceived Exertion
- OUH:
-
Oslo University Hospital
- DHI:
-
Dizziness Handicap Inventory
- PETS:
-
Problematic Experiences of Therapy Scale
References
Menon DK, Schwab K, Wright DW, Maas AI. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil. 2010;91:1637–40. https://doi.org/10.1016/j.apmr.2010.05.017.
Skandsen T, Nilsen TL, Einarsen C, Normann I, McDonagh D, Haberg AK, et al. Incidence of mild traumatic brain injury: a prospective hospital, emergency room and general practitioner-based study. Front Neurol. 2019;10:638. https://doi.org/10.3389/fneur.2019.00638.
Skandsen T, Einarsen CE, Normann I, Bjoralt S, Karlsen RH, McDonagh D, et al. The epidemiology of mild traumatic brain injury: the Trondheim MTBI follow-up study. Scand J Trauma, Resusc Emerg Med. 2018;26(1):34. https://doi.org/10.1186/s13049-018-0495-0.
Brazinova A, Rehorcikova V, Taylor MS, Buckova V, Majdan M, Psota M, et al. Epidemiology of traumatic brain injury in Europe: a living systematic review. J Neurotrauma. 2021;38(10):1411–40. https://doi.org/10.1089/neu.2015.4126.
Andelic N. The epidemiology of traumatic brain injury. Lancet Neurol. 2013;12(1):28–9. https://doi.org/10.1016/s1474-4422(12)70294-6.
Voormolen DC, Haagsma JA, Polinder S, Maas AIR, Steyerberg EW, Vulekovic P, et al. Post-concussion symptoms in complicated vs. Uncomplicated mild traumatic brain injury patients at three and six months post-injury: results from the CENTER-TBI study. J Clin Med. 2019;8:11. https://doi.org/10.3390/jcm8111921.
McIntyre M, Kempenaar A, Amiri M, Alavinia SM, Kumbhare D. The role of subsymptom threshold aerobic exercise for persistent concussion symptoms in patients with postconcussion syndrome: a systematic review. Am J Phys Med Rehabil. 2020;99(3):257–64. https://doi.org/10.1097/phm.0000000000001340.
Leddy JJ, Baker JG, Willer B. Active rehabilitation of concussion and post-concussion syndrome. Phys Med Rehabil Clin N Am. 2016;27(2):437–54. https://doi.org/10.1016/j.pmr.2015.12.003.
Ellis MJ, Leddy J, Cordingley D, Willer B. A physiological approach to assessment and rehabilitation of acute concussion in collegiate and professional athletes. Front Neurol. 2018;9:1115. https://doi.org/10.3389/fneur.2018.01115.
Silverberg ND, Iverson GL. Etiology of the post-concussion syndrome: physiogenesis and psychogenesis revisited. NeuroRehabilitation. 2011;29(4):317–29. https://doi.org/10.3233/nre-2011-0708.
Silverberg ND, Otamendi T. Advice to rest for more than 2 days after mild traumatic brain injury is associated with delayed return to productivity: a case-control study. Front Neurol. 2019;10:362. https://doi.org/10.3389/fneur.2019.00362.
Schneider KJ, Leddy JJ, Guskiewicz KM, Seifert T, McCrea M, Silverberg ND, et al. Rest and treatment/rehabilitation following sport-related concussion: a systematic review. Br J Sports Med. 2017;51(12):930–4. https://doi.org/10.1136/bjsports-2016-097475.
Voormolen DC, Polinder S, von Steinbuechel N, Vos PE, Cnossen MC, Haagsma JA. The association between post-concussion symptoms and health-related quality of life in patients with mild traumatic brain injury. Injury. 2019;50(5):1068–74. https://doi.org/10.1016/j.injury.2018.12.002.
Leddy JJ, Haider MN, Ellis M, Willer BS. Exercise is medicine for concussion. Curr Sports Med Rep. 2018;17(8):262–70. https://doi.org/10.1249/jsr.0000000000000505.
Kozlowski KF, Graham J, Leddy JJ, Devinney-Boymel L, Willer BS. Exercise intolerance in individuals with postconcussion syndrome. J Athl Train. 2013;48(5):627–35. https://doi.org/10.4085/1062-6050-48.5.02.
Ward SA, Whipp BJ. Exercise Physiology. In: Janes SM, editor. Encyclopedia of Respiratory Medicine (Second Edition). Academic Press; 2022. p. 196–208.
Leddy JJ, Haider MN, Ellis MJ, Mannix R, Darling SR, Freitas MS, et al. Early subthreshold aerobic exercise for sport-related concussion: a randomized clinical trial. JAMA Pediatr. 2019;173(4):319–25. https://doi.org/10.1001/jamapediatrics.2018.4397.
Willer BS, Haider MN, Bezherano I, Wilber CG, Mannix R, Kozlowski K, et al. Comparison of rest to aerobic exercise and placebo-like treatment of acute sport-related concussion in male and female adolescents. Arch Phys Med Rehabil. 2019;100(12):2267–75. https://doi.org/10.1016/j.apmr.2019.07.003.
Lal A, Kolakowsky-Hayner SA, Ghajar J, Balamane M. The effect of physical exercise after a concussion: a systematic review and meta-analysis. Am J Sports Med. 2018;46(3):743–52. https://doi.org/10.1177/0363546517706137.
Varner CE, Thompson C, de Wit K, Borgundvaag B, Houston R, McLeod S. A randomized trial comparing prescribed light exercise to standard management for emergency department patients with acute mild traumatic brain injury. Acad Emerg Med. 2021;28(5):493–501. https://doi.org/10.1111/acem.14215.
Leddy JJ, Willer B. Use of graded exercise testing in concussion and return-to-activity management. Curr Sports Med Rep. 2013;12(6):370–6. https://doi.org/10.1249/jsr.0000000000000008.
Leddy JJ, Baker JG, Kozlowski K, Bisson L, Willer B. Reliability of a graded exercise test for assessing recovery from concussion. Clin J Sport Med : J Can Acad Sport Med. 2011;21(2):89–94. https://doi.org/10.1097/JSM.0b013e3181fdc721.
Leddy JJ, Hinds AL, Miecznikowski J, Darling S, Matuszak J, Baker JG, et al. Safety and prognostic utility of provocative exercise testing in acutely concussed adolescents: a randomized trial. Clin J Sport Med : J Can Acad Sport Med. 2018;28(1):13–20. https://doi.org/10.1097/jsm.0000000000000431.
Mercier LJ, Fung TS, Harris AD, Dukelow SP, Debert CT. Improving symptom burden in adults with persistent post-concussive symptoms: a randomized aerobic exercise trial protocol. BMC Neurol. 2020;20(1):46. https://doi.org/10.1186/s12883-020-1622-x.
Skivington K, Matthews L, Simpson SA, Craig P, Baird J, Blazeby JM, et al. A new framework for developing and evaluating complex interventions: update of medical research council guidance. BMJ. 2021;374:n2061. https://doi.org/10.1136/bmj.n2061.
Chan AW, Tetzlaff JM, Gøtzsche PC, Altman DG, Mann H, Berlin JA, et al. SPIRIT 2013 explanation and elaboration: guidance for protocols of clinical trials. Bmj. 2013;346:e7586. https://doi.org/10.1136/bmj.e7586.
Calvert M, Kyte D, Mercieca-Bebber R, Slade A, Chan AW, King MT, et al. Guidelines for inclusion of patient-reported outcomes in clinical trial protocols: the SPIRIT-PRO extension. JAMA. 2018;319(5):483–94. https://doi.org/10.1001/jama.2017.21903.
Tverdal C, Aarhus M, Rønning P, Skaansar O, Skogen K, Andelic N, et al. Incidence of emergency neurosurgical TBI procedures: a population-based study. BMC Emerg Med. 2022;22(1):1. https://doi.org/10.1186/s12873-021-00561-w.
Carroll LJ, Cassidy JD, Holm L, Kraus J, Coronado VG, Injury WHOCCTFoMTB. Methodological issues and research recommendations for mild traumatic brain injury: the WHO collaborating centre task force on mild traumatic brain injury. J Rehabil Med. 2004;43(Suppl):113–25.
Riebe D, Franklin BA, Thompson PD, Garber CE, Whitfield GP, Magal M, et al. Updating ACSM’s recommendations for exercise preparticipation health screening. Med Sci Sports Exerc. 2015;47(11):2473–9. https://doi.org/10.1249/MSS.0000000000000664.
Haider MN, Leddy JJ, Wilber CG, Viera KB, Bezherano I, Wilkins KJ, et al. The predictive capacity of the buffalo concussion treadmill test after sport-related concussion in adolescents. Front Neurol. 2019;10:395. https://doi.org/10.3389/fneur.2019.00395.
Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc. 1982;14(5):377–81.
Nes BM, Janszky I, Wisloff U, Stoylen A, Karlsen T. Age-predicted maximal heart rate in healthy subjects: The HUNT fitness study. Scand J Med Sci Sports. 2013;23(6):697–704. https://doi.org/10.1111/j.1600-0838.2012.01445.x.
Howe EI, Fure SCR, Løvstad M, Enehaug H, Sagstad K, Hellstrøm T, et al. Effectiveness of combining compensatory cognitive training and vocational intervention vs. treatment as usual on return to work following mild-to-moderate traumatic brain injury: interim analysis at 3 and 6 month follow-up. Front Neurol. 2020;11:561400. https://doi.org/10.3389/fneur.2020.561400.
Eldridge SM, Chan CL, Campbell MJ, Bond CM, Hopewell S, Thabane L, et al. CONSORT 2010 statement: extension to randomised pilot and feasibility trials. Bmj. 2016;355:i5239. https://doi.org/10.1136/bmj.i5239.
King NS, Crawford S, Wenden FJ, Moss NE, Wade DT. The rivermead post concussion symptoms questionnaire: a measure of symptoms commonly experienced after head injury and its reliability. JNeurol. 1995;242(9):587–92.
WorkSafeBC Evidence-Based Practice Group, Martin CW. Post-Concussion Syndrome (PCS) - Validated Symptom Measurement Tools. BC Vancouver: In. Richmond; 2018.
Kosinski M, Bayliss MS, Bjorner JB, Ware JE Jr, Garber WH, Batenhorst A, et al. A six-item short-form survey for measuring headache impact: the HIT-6. Qual Life Res : Int J Qual Life Aspects Treat Care Rehabil. 2003;12(8):963–74. https://doi.org/10.1023/a:1026119331193.
Lerdal A, Wahl AK, Rustoen T, Hanestad BR, Moum T. Fatigue in the general population: a translation and test of the psychometric properties of the Norwegian version of the fatigue severity scale. Scand J Public Health. 2005;33(2):123–30.
Spitzer RL, Kroenke K, Williams JB, Lowe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092–7. https://doi.org/10.1001/archinte.166.10.1092.
Kroenke K, Spitzer RL, Williams JB, Löwe B. The patient health questionnaire somatic, anxiety, and depressive symptom scales: a systematic review. Gen Hosp Psychiatry. 2010;32(4):345–59. https://doi.org/10.1016/j.genhosppsych.2010.03.006.
Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Prim care eval mental disorders. Patient health questionnaire. JAMA. 1999;282:1737–44. https://doi.org/10.1001/jama.282.18.1737.
Lee PH, Macfarlane DJ, Lam TH, Stewart SM. Validity of the International Physical Activity Questionnaire Short Form (IPAQ-SF): a systematic review. Int J Behav Nutr Phys Act. 2011;8:115. https://doi.org/10.1186/1479-5868-8-115.
von Steinbuechel N, Petersen C, Bullinger M, Group Q. Assessment of health-related quality of life in persons after traumatic brain injury–development of the Qolibri, a specific measure. Acta Neurochir Suppl. 2005;93:43–9. https://doi.org/10.1007/3-211-27577-0_6.
von Steinbuechel N, Wilson L, Gibbons H, Muehlan H, Schmidt H, Schmidt S, et al. QOLIBRI overall scale: a brief index of health-related quality of life after traumatic brain injury. J Neurol Neurosurg Psychiatry. 2012;83(11):1041–7. https://doi.org/10.1136/jnnp-2012-302361.
Kirby S, Donovan-Hall M, Yardley L. Measuring barriers to adherence: validation of the problematic experiences of therapy scale. Disabil Rehabil. 2014;36(22):1924–9.
Jacobson GP, Newman CW. The development of the dizziness handicap inventory. Arch Otolaryngol Head Neck Surg. 1990;116(4):424–7. https://doi.org/10.1001/archotol.1990.01870040046011.
Evensen J, Soberg HL, Sveen U, Hestad KA, Bronken BA. The applicability of the Patient-Specific Functional Scale (PSFS) in rehabilitation for patients with Acquired Brain Injury (ABI) - a cohort study. J Multidiscip Healthc. 2020;13:1121–32. https://doi.org/10.2147/jmdh.S259151.
Moseng T, Tveter AT, Holm I, Dagfinrud H. Pasient-Spesifikk Funksjons Skala: Et nyttig verktøy for fysioterapeuter i primærhelsetjenesten. Fysioterapeuten. 2013;2:20–6.
Babic A, Tokalic R, AmílcarSilvaCunha J, Novak I, Suto J, Vidak M, et al. Assessments of attrition bias in Cochrane systematic reviews are highly inconsistent and thus hindering trial comparability. BMC Med Res Methodol. 2019;19(1):76. https://doi.org/10.1186/s12874-019-0717-9.
Fewtrell MS, Kennedy K, Singhal A, Martin RM, Ness A, Hadders-Algra M, et al. How much loss to follow-up is acceptable in long-term randomised trials and prospective studies? Arch Dis Child. 2008;93(6):458–61. https://doi.org/10.1136/adc.2007.127316.
Tamber AL, Wilhelmsen KT, Strand LI. Measurement properties of the Dizziness Handicap Inventory by cross-sectional and longitudinal designs. Health QualLife Outcomes. 2009;7:101. https://doi.org/10.1186/1477-7525-7-101.
Kleffelgaard I, Bruusgaard KA, Gjovaag T, Soberg HL. Subsymptomtrening for pasienter med treningsintoleranse etter lett traumatisk hodeskade – en pilotstudie. Fysioterapeuten. 2020;8:82-87.
El-Kotob R, Giangregorio LM. Pilot and feasibility studies in exercise, physical activity, or rehabilitation research. Pilot Feasibil Stud. 2018;4:137. https://doi.org/10.1186/s40814-018-0326-0.
Lancaster GA, Thabane L. Guidelines for reporting non-randomised pilot and feasibility studies. Pilot Feasibil Stud. 2019;5:114. https://doi.org/10.1186/s40814-019-0499-1.
Stallard N. Optimal sample sizes for phase II clinical trials and pilot studies. Stat Med. 2012;31(11–12):1031–42. https://doi.org/10.1002/sim.4357.
Acknowledgements
Not applicable.
Funding
Open access funding provided by University of Oslo (incl Oslo University Hospital). This clinical trial is founded by The Norwegian Fund for Post-Graduate Training in Physiotherapy.
Author information
Authors and Affiliations
Contributions
IK, HLS and NA developed the application with a preliminary study protocol based on the research at the University in Buffalo. LJVV is a phd-student in this project and has developed and conducted the feasibility study together with IK, HLS and MS. IK, HLS, MS, SES, NA and LJVV have contributed to the final study design and methods. All authors have contributed to drafting the final manuscript. All authors read and approved the final manuscript and consent to publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study is approved by the Regional Committee for Medical and Health Research Ethics South East Norway (#256109). All patients will receive written and oral information about the study from the treating physician or physiotherapist prior to the baseline assessment and provide their written informed consent prior to inclusion in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary file 1.
Feasibility study: Analysis of primary and secondary endpoints.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Valaas, LJ.V., Soberg, H.L., Rasmussen, M.S. et al. Sub-symptom threshold aerobic exercise for patients with persisting post-concussion symptoms and exercise intolerance after mild traumatic brain injury – a study protocol with a nested feasibility study for a randomized controlled trial. BMC Neurol 23, 179 (2023). https://doi.org/10.1186/s12883-023-03221-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-023-03221-7