
Abstract
Background
Proliferative glomerulonephritis with monoclonal immunoglobulin G (IgG) deposits (PGNMID) is a disease entity with nonorganized granular glomerular deposition with monoclonal proteins of both heavy and light chains. Dysproteinemia was observed in only 30% of the patients with PGNMID. We herein report a case of PGNMID with discrepancy between serum and glomerular deposits.
Case presentation
The patient was a 50-year-old man who had been followed at a local clinic due to hypertension, type 2 diabetes, hyperlipidemia, hyperuricemia, fatty liver, and obesity. Proteinuria had been noted five years previously, and he had been referred to a hematology department due to hyperproteinemia, high gamma globulin, and κ Bence-Jones protein (BJP) positivity one year previously. Bone marrow aspiration showed 5% plasma cells, and he was referred to the nephrology department to evaluate persistent proteinuria. He was hypertensive, and his estimated glomerular filtration rate was 54.2 ml/min/1.73 m2. His urinary protein level was 0.84 g/g⋅Cr. Urine and serum immunofixation showed BJP-κ type and IgG-κ type, respectively. Kidney biopsy showed an increase in mesangial cells and matrix without nodular lesions under a light microscope. Immunofluorescence microscopy showed granular deposits of IgG and C3 on the capillary wall and weak positivity for C1q. IgG3 was predominant among the IgG subclasses, and intraglomerular κ and λ staining was negative for κ and positive for λ. Direct fast scarlet staining was negative. Electron microscopy showed lumpy deposits without a fibrillar structure in the subepithelial area. Based on the above findings, a diagnosis of membranous nephropathy-type PGNMID was made. Since proteinuria increased gradually after three years of treatment with valsartan (40 mg, daily), oral prednisolone (30 mg, daily) was initiated, which led to decreased proteinuria. The dose of oral prednisolone was gradually tapered to 10 mg per day. At that time, proteinuria was 0.88 g/g⋅Cr. We found 204 cases in 81 articles in the PubMed database, among which 8 showed discrepancy in the heavy and/or light chains between serum and kidney.
Conclusions
We experienced a case of membranous nephropathy-type PGNMID with discrepancy in light chains between serum and kidney that was successfully treated with oral prednisolone.
Similar content being viewed by others


Background
Proliferative glomerulonephritis with monoclonal immunoglobulin G (IgG) deposits (PGNMID) was first reported as a disease entity with nonorganized granular glomerular deposition with monoclonal proteins of both heavy and light chains other than type 1 cryoglobulinemic glomerulonephritis, Randall type light and heavy chain deposition disease, immunotactoid glomerulonephritis, and fibrillary glomerulonephritis [1].
Dysproteinemia was observed in only 30% of the patients with PGNMID, and glomerular deposition with IgG3 was the most common finding [2]. After recognizing this disease entity, cases accumulated, some of which showed discrepancy between serum and glomerular deposits [3,4,5,6,7,8]. We herein report an additional case of PGNMID with discrepancy between serum and glomerular deposits.
Case presentation
The patient was a 50-year-old man who had been followed up at a local clinic due to hypertension, type 2 diabetes, hyperlipidemia, hyperuricemia, fatty liver, and obesity. His medications included valsartan, voglibose, bezafibrate, and febuxostat. Although proteinuria was noted at an annual health checkup five years previously, proteinuria was not detected at the subsequent health checkup. Then, hyperproteinemia (9 g/dl), a high gamma globulin level, and κ Bence-Jones protein (BJP) positivity were noted, and he was referred to the hematology department of Mie University Hospital one year previously. Bone marrow aspiration showed 5% plasma cells, which did not lead to a diagnosis of multiple myeloma. Since he had persistent proteinuria, he was referred to the nephrology department and admitted for close examination.
His height was 170 cm, his body weight was 97.5 kg, and his body mass index was 33.7. His blood pressure was 151/103 mmHg. Although a physical examination did not reveal any obvious enlarged lymph nodes or hepatosplenomegaly, computed tomography (CT) showed mildly enlarged cervical, supraclavicular, axillary, mediastinal, para-aorta, hepatic portal, and bilateral inguinal lymph nodes, splenomegaly, and fatty liver. Positron emission tomography (PET)-CT showed mild accumulation of fluorodeoxyglucose in the cervical, supraclavicular, axillary, and inguinal lymph nodes. His laboratory data are shown in Table 1. His urinary protein level was 0.84 g/g⋅Cr without occult blood, and urine immunofixation showed BJP-κ type. Serum immunofixation showed IgG-κ type, and the blood immunoglobulin free light chain κ/λ ratio was also elevated (Table 1).
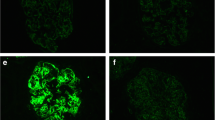
Kidney biopsy showed 24 glomeruli, of which 2 showed global sclerosis, 2 showed segmental sclerosis, and 1 collapsed. Tubulointerstitial damage was observed in 10–20% of patients. There was an increase in mesangial cells and matrix without nodular lesions under light microscopy (Fig. 1). No obvious spike formation was observed under high magnification with periodic acid methenamine silver staining (Fig. 1). An immunofluorescence study showed granular deposits of IgG and C3 on the capillary wall and weak positivity for C1q (Fig. 2a). IgG3 was predominant among the IgG subclasses, and intraglomerular κ and λ staining was negative for κ and positive for λ (Fig. 2b). Direct fast scarlet staining was negative. Electron microscopy revealed lumpy deposits without a fibrillar structure in the subepithelial area (Fig. 3).

Light microscopy findings. There was an increase in mesangial cells and matrix without nodular lesions in periodic acid-Schiff staining (upper panel) or periodic acid-methenamine silver (PAM) staining (middle panel). There was no obvious spike formation under high magnification with PAM staining (lower panel). Bars = 50 μm

a Immunofluorescence findings. The immunofluorescence study showed granular deposits of immunoglobulin G (IgG) and complement 3 on the capillary wall and weak positivity for complement 1q. b IgG3 was predominant among the IgG subclasses, and intraglomerular κ and λ staining were negative for κ and positive for λ. Bars = 100 μm

Electron microscopy findings. Electron microscopy showed electron-dense deposits (EDD) in the subepithelial area (arrowheads, upper panel). High magnification of the EDD showed granular nonorganized deposits without a fibrillar structure (lower panel). Bars = 2 μm
Based on the above findings, a diagnosis of membranous nephropathy-type PGNMID was made. Considering that hypertension and obesity-related hyperfiltration may have contributed to the urinary protein, he was treated with sodium restriction and body weight loss in addition to valsartan (40 mg, daily) (Fig. 4). Since his proteinuria increased gradually over the three years, oral prednisolone (30 mg, daily) was initiated, which led to a decrease in proteinuria. Valsartan was changed to azilsartan (40 mg, daily), and teneligliptin (20 mg, daily) was initiated. Oral prednisolone was gradually tapered to a dose of 10 mg per day, and his proteinuria was 0.88 g/g⋅Cr with the use of azilsartan (40 mg daily, canagliflozin (100 mg, daily), and esaxerenone (1.25 mg, daily).

The clinical course. eGFR, estimated glomerular filtration rate
Discussion and conclusions
We experienced a case of PGNMID that showed a discrepancy in light chains between serum and kidney. While the present case had IgG-κ and BJP-κ in serum and urine, a bone marrow examination did not lead to a diagnosis of multiple myeloma. Kidney biopsy showed increased mesangial cells and matrix on light microscopy, and an immunofluorescence study showed granular deposits of IgG3 and λ light chain on the capillary wall. Direct first scarlet staining was negative, and electron microscopy showed granular electron-dense deposits in the subepithelial area. Based on the above findings, a diagnosis of membranous nephropathy-type PGNMID was made.
While IgG-κ was identified in the serum of the present case and IgG1 was predominant in the serum, the glomerular deposition was composed of IgG3-λ. Therefore, we searched the PubMed database using the term “glomerulonephritis with monoclonal IgG deposits” or “proliferative glomerulonephritis with monoclonal immunoglobulin deposits”. Among 204 cases in 81 articles, 8 cases showed discrepancy in heavy and/or light chains between serum and kidney (Table 2) [3,4,5,6,7,8]. Glomerular deposition of IgG, IgA, and IgM was observed in 5, 3, and 1 cases, respectively. Discrepancy of the heavy chain was reported in 4 cases. Discrepancy of the light chain was reported in 7 cases. Regarding the histological pattern, mesangial proliferative glomerulonephritis, membranoproliferative glomerulonephritis, and membranous nephropathy were found in 4, 3, and 2 cases, respectively. Although the significance of discrepancy in κ and λ is not clear, IgG3 is characterized by a strong positive charge and large molecular weight, which might explain its affinity to the glomeruli [2]. In the present case, IgG1-κ was positive in serum, and IgG3-λ was positive in the glomeruli, suggesting that the affinity of IgG3-λ to the glomeruli caused a discrepancy between serum and kidney.
Since there is no definitive treatment for PGNMID [9], renin–angiotensin system inhibitors were considered to be a reasonable treatment for the present case. Initially, the patient showed a decrease in urinary protein with valsartan alone; however, his proteinuria gradually increased, and kidney dysfunction also appeared. Since a previous report showed successful treatment with oral prednisolone in a case of membranous nephropathy-type PGNMID with IgG2-κ deposition [10], oral prednisolone was started, and proteinuria decreased in the present case. A clone-directed approach will be needed for cases with aggravation of kidney function and proteinuria in the future [7, 11].
The anti-nuclear antibody (ANA) and anti-SS-A antibody levels were high in this case (2560 times and 240 times, respectively). According to the 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for systemic lupus erythematosus (SLE) [12], the present case had urinary protein > 0.5 g/day (4 points) and low C4 (3 points) but no other systemic symptoms, blood findings, or abnormal findings in the skin, mucosa, or musculoskeletal system; thus, a diagnosis of SLE was not made. While the C4 level was low, we did not investigate the presence of immune complex in serum by a C1q binding assay or a monoclonal Rheumatoid factor assay in the present case. There were also no findings of dryness of the eyes and no abnormalities of the salivary or parotid glands; thus, a definitive diagnosis of Sjögren’s syndrome could not be made. However, hypergammaglobulinemia might imply the subclinical status of these autoimmune diseases, and there was a possibility of the contribution of polyclonal gammopathy to the discrepancy of this serum and glomerular light chain dominancy. As there was no description of the presence of hypergammaglobulinemia in the other eight cases in Table 2, we could not elucidate whether there was a common feature of hypergammaglobulinemia that was related to the discrepancy between serum and kidney. There might be a monoclonal gammopathy of undetermined significance with IgG1-κ, which did not contribute to kidney injury, and a monoclonal gammopathy of renal significance with IgG3-λ, which was specifically deposited in the kidney in the present case. The presence of two B-cell clones producing different M proteins was difficult to prove in the flow cytometry analysis of the bone marrow of the present case. While the majority were IgG1-κ-producing B-cell clones in the present case, a small amount of IgG3-λ B-cell clones were present, which could not be detected in serum or urine and which tended to be deposited in the kidney at the same time [13]. This is compatible with a previous study that reported that the rate of detectable monoclonal immunoglobulins in PGNMID was low at 30–32% [14].
The present case was diagnosed with membranous nephropathy-type PGNMID, which was almost synonymous with nonorganized and non-Randall-type monoclonal immunoglobulin deposition disease associated with membranous features. While there was a report of a case with membranous nephropathy with monoclonal IgM-λ deposition that showed THSD7A positivity [15], we could not search for antigens associated with membranous nephropathy (e.g., PLA2R, THSD7A, and NELL1) due to a lack of samples in the present case; therefore, it was impossible to distinguish the present case from primary membranous nephropathy with monoclonal immunoglobulin deposits.
In conclusion, we experienced a case of membranous nephropathy-type PGNMID with a discrepancy in light chains between serum and kidney.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACR:
-
American College of Rheumatology
- ANA:
-
Anti-nuclear antibodies
- BJP:
-
Bence-Jones protein
- CT:
-
Computed tomography
- EULAR:
-
European League Against Rheumatism
- IgG:
-
Immunoglobulin G
- PET:
-
Positron emission tomography
- PGNMID:
-
Proliferative glomerulonephritis with monoclonal IgG deposits
- SLE:
-
Systemic lupus erythematosus
References
Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, Appel GB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65(1):85–96. https://doi.org/10.1111/j.1523-1755.2004.00365.x.
Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20(9):2055–64. https://doi.org/10.1681/ASN.2009010110.
Redondo-Pachón MD, Enríquez R, Sirvent AE, Andrada E, García-Del Moral R, Millán I, et al. Proliferative glomerulonephritis with monoclonal IgG deposits on multiple myeloma. Nefrologia. 2012;32:846–8.
Batal I, Bijol V, Schlossman RL, Rennke HG. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits in a kidney allograft. Am J Kidney Dis. 2014;63(2):318–23. https://doi.org/10.1053/j.ajkd.2013.07.015.
Vignon M, Cohen C, Faguer S, Noel LH, Guilbeau C, Rabant M, et al. The clinicopathologic characteristics of kidney diseases related to monotypic IgA deposits. Kidney Int. 2017;91(3):720–8. https://doi.org/10.1016/j.kint.2016.10.026.
Shimohata H, Ohgi K, Maruyama H, Miyamoto Y, Takayashu M, Hirayama K, et al. A case of proliferative glomerulonephritis with monoclonal IgG deposits that showed predominantly membranous features. Case Rep Nephrol. 2017;2017:1027376.
Gumber R, Cohen JB, Palmer MB, Kobrin SM, Vogl DT, Wasserstein AG, et al. A clone-directed approach may improve diagnosis and treatment of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Kidney Int. 2018;94(1):199–205. https://doi.org/10.1016/j.kint.2018.02.020.
Rosenstock JL, Vynnyk M, DeVita MV, D’Agati VD. Two cases of proliferative glomerulonephritis with monoclonal IgG deposits treated with renin angiotensin inhibition alone with long-term follow-up. Kidney Int Rep. 2021;6:2218–22.
Bridoux F, Javaugue V, Nasr SH, Leung N. Proliferative glomerulonephritis with monoclonal immunoglobulin deposits: a nephrologist perspective. Nephrol Dial Transplant. 2021;36(2):208–15. https://doi.org/10.1093/ndt/gfz176.
Ohashi R, Sakai Y, Otsuka T, Ohno D, Masuda Y, Murasawa T, et al. Proliferative glomerulonephritis with monoclonal IgG2κ deposit successfully treated with steroids: a case report and review of the literature. CEN Case Rep. 2013;2(2):197–203. https://doi.org/10.1007/s13730-013-0064-3.
Guiard E, Karras A, Plaisier E, Van Duong JP, Fakhouri F, Rougier JP, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. 2011;6:1609–16.
Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic Lupus Erythematosus. Arthritis Rheumatol. 2019;71:1400–12.
Bridoux F, Leung N, Hutchison CA, Touchard G, Sethi S, Fermand JP, et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015;87(4):698–711. https://doi.org/10.1038/ki.2014.408.
Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D’Agati VD, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45–59. https://doi.org/10.1038/s41581-018-0077-4.
Hirose G, Uchida T, Kojima A, Sugisaki K, Yamada M, Nagase Y, et al. Membranous Nephropathy with Monoclonal IgM Lambda deposits in a patient with IgM Monoclonal Gammopathy: a Case Report. Front Med (Lausanne). 2021;8:608741.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SM, KK, FT, MM, and DT participated in the acquisition of clinical data. SM, KK, YS, RS, YH, TM, IT, and KD carried out the analysis of the patient’s clinical course and data interpretation. SM and KK wrote a draft of the manuscript, and YS, FT, MM, DT, RS, YH, TM, IT, and KD revised it critically. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for the publication of this case report. A copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare no conflicts of interest in association with the present study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Miura, S., Katayama, K., Sugimoto, Y. et al. Discordance of light chain isotypes between serum and glomerular deposits in proliferative glomerulonephritis with monoclonal IgG deposits: a case report and review of the literature. BMC Nephrol 24, 199 (2023). https://doi.org/10.1186/s12882-023-03256-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-023-03256-5