
Abstract
Background
Proliferative glomerulonephritis with monoclonal immunoglobulin G (IgG) deposits (PGNMID) is a rare monoclonal gammopathy of renal significance with dense deposits of monoclonal immunoglobulin.
Case presentation
We report a 78-year-old Japanese male patient with mild proteinuria and lower extremity edema. Monoclonal immunoglobulin could not be identified in his serum or urine. Although his bone marrow biopsy was negative, renal biopsy found features of membranoproliferative glomerulonephritis (MPGN) with deposition of monoclonal IgG2 kappa. Electron microscopy examination revealed non-organized electron-dense deposits in the subepithelial, and subendothelial mesangial regions. Steroid monotherapy was performed after diagnosis of PGNMID but complete remission was not achieved.
Conclusion
PGNMID with IgG3 kappa deposits is the most common in cases with the histological feature of MPGN. There are few cases of PGNMID with IgG2 kappa deposits exhibiting MPGN. This report describes a very rare case of PGNMID with the histological feature of MPGN.
Similar content being viewed by others

Background
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) is a rare disease that is regarded as one subtype of monoclonal gammopathy of renal significance [1]. It is reported that the renal biopsy incidence of PGNMID is only about 0.17% [1]. Clinically, patients with proteinuria and microscopic hematuria gradually develop renal dysfunction. They have renal pathology including endocapillary proliferative, membranoproliferative or membranous features with positive staining of glomerular IgG or IgA deposits, which are restricted to a single IgG or IgA subclass [1, 2]. The efficacy of immunosuppressive agents, such as steroids, cyclophosphamide, cyclosporine, mycophenolate, bortezomib, rituximab or daratumumab, for treatment of PGNMID has not been established [3,4,5].
Here, we present a case of PGNMID that was treated with steroid monotherapy. The biopsy revealed PGNMID with features of membranoproliferative glomerulonephritis (MPGN), characterized by the presence of monoclonal IgG2 kappa and non-organized electron-dense deposits (EDDs), which are located in the subepithelial, and subendothelial regions. This report describes a very rare case of PGNMID with IgG2 kappa deposits exhibiting MPGN.
Case presentation
A 78-year old man with type 2 diabetes mellitus and hypertension presented with mild proteinuria, lower extremity edema, and worsening renal function. His serum creatinine level was 1.22 mg/dL just before referral to our hospital. His type 2 diabetes mellitus was well controlled for several years (hemoglobin A1c 6.2–7.4%) and there was no family history of kidney disease. On admission to our hospital, his blood pressure was 132/48 mmHg, pulse rate 78 beats/min and body temperature 36.9 °C. Physical examination revealed no significant systolic heart murmur on auscultation. Examination of the lungs, abdomen, and central and peripheral nervous systems found no abnormalities. However, mild edema was observed in the lower extremities. The patient’s laboratory data on admission are summarized in Table 1. Urinalysis showed markedly elevated proteinuria (protein excretion, 6.37 g/day) and hematuria (10–19 red blood cells/high-powered field). Serum albumin level was 3.1 g/dL, and serum creatinine level was 1.68 mg/dL (reference range, 0.65–1.07 mg/dL), with estimated glomerular filtration rate of 56 mL/min/1.73 m2. White blood cell count was 8.62 × 103 cells/μL (reference range, 3.3 × 103–8.6 × 103 cells/μL), hemoglobin level was 10.5 g/dL (reference range, 13.7–16.8 g/dL), and platelet count was 22.4 × 104 cells/μL (reference range, 15.8 × 104–34.8 × 104 cells/μL). Serum IgG level was 1120 mg/dL (reference range, 861–1747 mg/dL), IgA level was 222 mg/dL (reference range, 93–393 mg/dL), IgM level was 28 mg/dL (reference range, 33–183 mg/dL), and IgE level was 539 U/mL (reference range, 0–360 U/mL). Both serum free kappa (64.7 mg/dL) and lambda light (46.9 mg/dL) chain levels were mildly elevated (reference range of free kappa level and free lambda light levels, 3.3–19.4 and 5.7–26.3 mg/dL, respectively) and there was no deviation. M-protein on serum immunofixation electrophoresis was negative. Bence–Jones protein on urine immunofixation electrophoresis was negative. Plasma C3 level was 95 mg/dL (reference range, 73–138 mg/dL), and C4 level was 27 mg/dL (reference range, 11–31 mg/dL). Hepatitis B surface antigen, and hepatitis C virus, human immunodeficiency virus, and human T-cell leukemia virus antibodies were all negative. The patient was positive for anti-nuclear antibodies but negative for anti-neutrophil cytoplasmic, anti-glomerular basement membrane and anti-DNA antibodies, and cryoglobulin (Table 1). His bone marrow biopsy exhibited absence of clonal B cells or plasma cells.
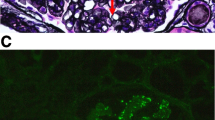
Based on the above clinical findings, percutaneous renal biopsy was performed to determine the pathological characteristics of the renal dysfunction. The biopsied sample consisted of a renal cortex containing 14 glomeruli, all of which were segmentally or globally non-sclerotic. The histological pattern was predominantly MPGN characterized by diffuse and global double-contoured glomerular capillary walls with mesangial cell interposition and mesangial expansion by increased mesangial cell number and matrix. Moreover, it showed endocapillary hypercellularity (Fig. 1). Masson trichrome staining showed some tubulointerstitial damage (data not shown). Direct first scarlet staining revealed no amyloid deposition (data not shown). Immunofluorescence produced strong glomerular staining of IgG and C3 globally in the peripheral capillary loops and segmentally in the mesangial areas (Fig. 2). The deposited IgG was composed only of subclass IgG2 kappa light chain (Figs. 2 and 3). Electron microscopy revealed that EDDs were located in the subepithelial and subendothelial regions. Non-organized structures were readily identified within the EDDs (Fig. 4). No EDDs were detected in the tubulointerstitium and arterial walls (data not shown). Based on the above histological and immunohistological findings, we diagnosed this case as PGNMID with monoclonal IgG2 kappa deposits exhibiting MPGN features.

Light microscopy findings (PAS stain; PAM stain, 400x). Light microscopy Olympus BX53, OLYMPUS DP73 camera, and cellSense (Olympus Corporation, Center Valley, PA, USA) was used to capture the images. The images were obtained with eyepiece at 10X magnification and objective at 40X. No enhancement of the images was performed. The measured resolution was 4800 × 3600. The histological pattern was predominantly membranoproliferative glomerulonephritis characterized by diffuse and global double-contoured glomerular capillary walls (red arrow) with mesangial cell interposition and mesangial expansion by increased mesangial cell number and matrix. Moreover, it showed endocapillary hypercellularity (black arrow)

Immunofluorescence analysis was performed using fluorescein-isothiocyanate-conjugated antibody against IgG, IgA, IgM, C3, C1q, fibrinogen, kappa chain, lambda light chain or nephritis-associated plasmin receptor (NAPlr). Fine granular staining of IgG (2+), C3 (2+) and kappa chain (2+) was identified, along the glomerular capillary walls and in the mesangial areas, while IgA, IgM, C1q, fibrinogen, lambda light chain, and NAPlr were all negative (400x)

Immunofluorescence analysis was performed using primary antibodies against IgG1, IgG2, IgG3or IgG4 and secondary fluorescein-isothiocyanate-conjugated antibody. Fine granular staining of IgG 2 (2+) was identified, along the glomerular capillary walls and in the mesangial areas, while the other IgG subclasses (IgG1, IgG3 and IgG4) were all negative (400x)

Electron microscopy revealed electron-dense deposits (EDDs; black arrows) located in the subepithelial (red arrow) and subendothelial (black arrow) regions. Non-organized structures could readily be identified within the EDDs
Steroid pulse therapy (methylprednisolone 500 mg/day for 3 days) and subsequent oral administration of 40 mg/day (0.6 mg/kg) prednisolone were performed after diagnosis of PGNMID with monoclonal IgG2 kappa deposits. The prednisolone dose was gradually reduced to 15 mg/day over 6 months. Although proteinuria and serum creatinine level were not changed, serum albumin level gradually increased (Fig. 5).

Clinical course of the patient. Abbreviations: Alb, albumin; Cr, creatinine; mPSL, methylprednisolone; PSL, prednisolone. PSL dose given in mg/day
Discussion and conclusions
To our knowledge, this is a rare case of PGNMID with monoclonal IgG2 kappa deposit, exhibiting MPGN features. Renal biopsy revealed MPGN characterized by diffuse and global double-contoured glomerular capillary walls with mesangial cell interposition and mesangial expansion by increased mesangial cell number and matrix. Immunofluorescence revealed monoclonal IgG2 kappa deposits within the glomeruli, which was suggestive of PGNMID. Electron microscopy analysis showed non-organized EDDs in the subepithelial, and subendothelial regions.
Monoclonal gammopathy of renal significance (MGRS) is defined as hematological disorders with kidney disease [3]. Importantly, the B-cell or plasma-cell disorders of patients with MGRS do not demand any current hematological criteria for multiple myeloma and B-cell lymphoproliferative disorders. MGRS is commonly the consequence of the renal deposition of monoclonal immunoglobulins or their components either directly or indirectly. PGNMID is a rare disease, which was reported first by Nasr et al. [1]. They described three main characteristics of PGNMID: (1) endocapillary proliferative, membranoproliferative or membranous glomerulonephritis with glomerular IgG deposits, which are restricted to a single IgG subclass (IgG1–4) and a single light chain subclass (kappa or lambda); (2) the presence of non-orginized deposits by electron microscopy; and (3) exclusion of cryoglobulinemia [1]. Thus, PGNMID is classified as one subtype of MGRS [3]. Our case demanded the above three features. However, neither serum immunoelectrophoresis nor urine electrophoresis suggested monoclonal gammopathy. Moreover, the bone marrow biopsy exhibited absence of clonal plasma cells. Actually, approximately 70% of cases do not have detectable blood or bone marrow monoclonal immunoglobulins [4]. Thus, many PGNMID cases might not detect monoclonal immunoglobulins. A potential limitation about this is that a clonal small population could not be detected by the currently used techniques possibly. Another possible explanation is that the clone is located in the extra-medullary lymphoid tissue or organs [6].
The most common histological pattern of PGNMID is MPGN pattern, characterized by diffuse and global double-contoured glomerular capillary walls with mesangial deposits. The second most common type is endocapillary proliferative glomerulonephritis pattern, which is characterized by endocapillary hypercellularity and active inflammatory cell infiltration [1]. IgG3 kappa is the most common type of immunoglobulin in cases with the histological pattern of MPGN. Some reports show that about 50% of PGNMID patients have IgG3 deposition, and about 30% have IgG1 deposition [1, 7, 8]. Interestingly, there are few cases of PGNMID with IgG2 kappa exhibiting MPGN features, although there is one report of PGNMID with IgG2 lambda exhibiting MPGN features and another about PGNMID with IgG2 kappa exhibiting membranous nephropathy [9, 10]. Moreover, Bhutani et al. reported that PGNMID with IgG3 tended to have MPGN pattern, whereas PGNMID with IgG1 tended to show MN pattern [6, 11]. Regarding PGNMID with IgG4, there was one report about MN pattern [12]. The management of PGNMID is not well established. Thus, the treatment is based on some factors, including the presence or absence of monoclonal immunoglobulins and the risk for progression of kidney disease. Many patients have been treated with renin–angiotensin system (RAS) inhibition alone or with immunosuppressive agents including steroids, cyclophosphamide, cyclosporine, mycophenolate, bortezomib, rituximab or daratumumab, with variable success rates [3,4,5,5, 13]. Nasr et al. reported that only four of 32 patients with PGNMID achieved complete remission (reduction of proteinuria to < 500 mg/day). Many patients did not show significant recovery, although various kinds of therapies were administered. Only one of the six patients treated with steroid monotherapy had complete remission, and the others had incomplete remission, persistent renal insufficient or end-stage renal disease [1]. In our case, proteinuria and serum creatinine level were not changed, even though steroid monotherapy were administered. Additional therapies, including other immunosuppressive agents, might be necessary.
This is believed to be the rare case of proliferative glomerulonephritis with monoclonal IgG2 kappa deposition exhibiting MPGN features. Further studies on a larger number of PGNMID cases are required to confirm the association between IgG subclasses and clinical outcomes.
Availability of data and materials
All data generated or analysed during this study were obtained from the corresponding author on reasonable request.
References
Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20:2055–64.
Kusunoki Y, Namba-Hamano T, Kakimoto T, et al. A case of proliferative glomerulonephritis with immunoglobulin A1-lambda deposits successfully treated by chemotherapy. CEN Case Rep. 2020;9:326–32.
Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the international kidney and monoclonal Gammopathy research group. Nat Rev Nephrol. 2019;15:45–59.
Noto R, Kamiura N, Ono Y, Tabata S, et al. Successful treatment with bortezomib and dexamethasone for proliferative glomerulonephritis with monoclonal IgG deposits in multiple myeloma: a case report. BMC Nephrol. 2017;18:127.
Yu XJ, Wang MJ, Yong ZH, et al. Proliferative glomerulonephritis with monoclonal IgG3λ deposits: a case report of a rare cause of monoclonal gammopathy of renal significance. Kidney Med. 2019;1:221–5.
Bhutani G, Nasr SH, Said SM, et al. Hematologic characteristics of proliferative glomerulonephritides with nonorganized monoclonal immunoglobulin deposits. Mayo Clin Proc. 2015;90:587–96.
Nasr SH, Markowitz GS, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85–96.
Guiard E, Karras A, Plaisier E, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. 2011;6:1609–16.
Sumida K, Ubara Y, Marui Y, et al. Recurrent proliferative glomerulonephritis with monoclonal IgG deposits of IgG2λ subtype in a transplanted kidney: a case report. Am J Kidney. 2013;62:587–90.
Ohashi R, Sakai Y, Otsuka T, et al. Proliferative glomerulonephritis with monoclonal IgG2κ deposit successfully treated with steroids: a case report and review of the literature. CEN Case Rep. 2013;2:197–203.
Li M, Xu G. An update of proliferative glomerulonephritis with monoclonal immunoglobulin deposits. Clin Kidney J. 2021;15:1041–8.
Omokawa A, Komatsuda A, Hirokawa M, et al. Membranous nephropathy with monoclonal IgG4 deposits and associated IgG4-related lung disease. Clin Kidney J. 2014;7:475–8.
Zand L, Rajkumar SV, Leung N, et al. Safety and efficacy of daratumumab in patients with proliferative GN with monoclonal immunoglobulin deposits. J Am Soc Nephrol. 2021;32:1163–73.
Acknowledgements
Not applicable.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
DI collected the clinical data and were involved in the clinical care of the patient. DI and MT carried out the hematologic studies and carried out the treatment. DI, AT and T M made the pathological diagnosis. DI, YS1, YS2, YS3, and MT contributed an interpretation of clinical course. YS1 corresponded to Yuriko Shiozaki. YS2 corresponded to Yoshitaka Shimizu. YS3 corresponded to Yumiko Suzuki. DI and TM wrote the manuscript. AT supervised the manuscript. All of the authors have contributed to the preparation of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the editor of this journal.
Competing interests
All the authors have declared no competing interest exists.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ito, D., Shiozaki, Y., Shimizu, Y. et al. A rare case of proliferative glomerulonephritis with monoclonal IgG2 kappa deposit: a case report. BMC Nephrol 23, 396 (2022). https://doi.org/10.1186/s12882-022-03029-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-022-03029-6