
Abstract
Background
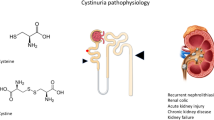
Cystinuria is an inherited metabolic disease that is caused by defects in two genes, SLC3A1 and SLC7A9, which result in a renal reabsorptive defect of cystine and other dibasic amino acids, including ornithine, arginine, and lysine. Patients usually present with recurrent renal calculi and may develop renal impairment. Medical management includes high fluid intake and chelating agents. To the best of our knowledge, this is the first study describing cystinuria in Saudi Arabia.
Methods
A retrospective chart review for cystinuria patients from the genetic and nephrology divisions between 2010 to 2015. All patients were investigated, diagnosed and treated at King Abdulaziz Medical City in Saudi Arabia.
Results
Eight patients were identified from five unrelated families. The age of onset ranged from birth to 14 years. The female to male ratio was 1.7:1. Two new variants in the SLC3A1 and SLC9A7 genes were discovered. All of the detected mutations were missense variants in three different exons, such as c.1711 T > A (p.Cys571Ser) (exon 10), c.1166C > T p.Thr389Met (exon 11) and c.1400 T > A p.Met467Lys (exon 8). Additionally, 37.5% of our patients developed arterial hypertension and 25% had urinary tract infection, but none had renal impairment. No significant clinical differences were detected in this study between type A (SLC3A1 variants) and type B cystinuria (SLC7A9 variant). Two cases were diagnosed based on clinical information, biochemical testing and a positive family history as all of the molecular testing for cystinuria was negative.
Conclusion
Cystinuria has wide genetic heterogeneity with a poor genotype/phenotype correlation. Negative molecular investigations should not rule out the disease if clinical and biochemical investigations support the diagnosis. A larger data registry is essential to better describe the cystinuria genotype/phenotype in Saudi Arabia.
Similar content being viewed by others


Background
Cystinuria continues to be one of the most challenging stone diseases [1], which has a recurrence rate up to 60%. It is associated with progressive renal impairment [2]. Cystinuria is one of four genetic metabolic diseases discovered by Carrod in 1908 [3]. A recent study published from our center predicted the incidence of cystinuria to be 4.5/100,000 live births [4].
Thus far, cystinuria is thought to be caused by defects in two genes, SLC3A1 and SLC7A9. SLC3A1 (MIM #104614) is located on chromosome 2 (2P16.3) and encodes the heavy subunit of the renal amino acid transporter (rBAT) needed to localize the transporter to the plasma membrane. The SLC7A9 gene (CSNU3, MIM #604144) is located on chromosome 19 (19q13.1) and encodes the light subunit of the renal amino acid transporter (b0,+AT), which compromises the catalytic, transporting component. The Rbat/B0,+AT is linked by a disulfide bridge [5–7]. The mode of transmission seems to be autosomal recessive or autosomal dominant with incomplete penetrance [8, 9].
Amino acid transport defects cause hyper-excretion and insolubility of cystine and other dibasic amino acids as well as precipitation of cystine at the physiological pH of urine, which results in recurrent cystine renal calculi, obstructive uropathy, hypertension, infection and rarely renal failure [10]. More than 50% of cystinuric patients develop cystine urolithiasis, and 75% of them have bilateral stones. Recurrent stone formation requires repeated urological interventions [11, 12].
The diagnosis of cystinuria is achieved by functional, biochemical or molecular investigations. Functionally, patients excreting cystine more than 120 mmol/mol creatinine in 24 h urine are defined as having cystinuria [3]. Biochemically, cystinuria is divided into two subtypes based on the amount of cystine excreted in the urine. For type 1 cystinuria, which has an autosomal recessive mode of transmission, heterozygotes have normal amino acid excretion. On the other hand, for non-type 1 cystinuria, heterozygotes have cystine hyper-excretion, indicating an autosomal dominant mode of transmission with incomplete penetrance [9]. Molecularly, mutations in the SLC3A1 gene cause type A cystinuria, while type B is due to SLC7A9 gene mutations. A possible third type, AB, has mutations in both genes [8]. Although it was proposed that SLC3A1 mutations (type A) cause type 1 cystinuria and SLC7A9 mutations (type B) cause non-type 1 cystinuria, an increasing number of SLC3A1 variants were discovered to cause cystine and other dibasic hyper-aminoaciduria in carriers in addition to several point mutations in SLC7A9, resulting in silent heterozygotes [10].
The treatment success in cystinuria patients depends on full compliance with medical therapy, including urine alkalization with potassium citrate and dietary sodium restriction. High fluid intake has been shown to decrease urinary stone formation in up to 67% of patients with cystinuria, achieving a urine specific gravity of 1.010. The most effective chelating agent for cystinuria is oral thiol (β,β-dimethylcystine), which forms mixed disulfides with cystine, dissolving and reducing new cystine stone formation [13].
In this study, we described the clinical phenotype and genotype of eight Saudi patients with cystinuria. To the best of our knowledge, this is the first study in the literature describing the clinical, biochemical and molecular characteristics of cystinuria in Saudi Arabia.
Methods
A retrospective chart review of eight Saudi cystinuric patients from five unrelated families between 2010 to 2015. All patients were diagnosed and treated at King Abdulaziz Medical City in Saudi Arabia under the care of a multidisciplinary team, including nephrologists and geneticists.
The diagnosis was clinically established based on recurrent cystine renal calculi formation that started in childhood. Other biochemical, radiological and molecular investigations were requested to confirm the diagnosis. Investigations that were performed include abdominal X-Ray, ultrasound, abdominal CT, stone analysis, urinary amino acid study and molecular genetic testing. Amino acids in urine were measured using early morning fresh urine samples and 24-h urine. DNA sequencing analysis, coding sequences of the SLC3A1 and SLC7A9 genes and mutational analysis were performed at referenced commercial labs. The SLC3A1(NM_000341) and SLC7A9 (NM_014270) genes were analyzed by PCR and sequencing of both DNA strands of the coding introns and highly conserved exon-intron splice junctions.
Results
Clinical data (Table 1)
All patients were products of consanguineous parents. They presented with renal stone related signs and symptoms; five patients presented with bilateral renal calculi and three had unilateral stones. The ages of onset were variable, ranging from birth to 14 years (mean 6.3 years). The current age of the oldest patient is 24 years. The female to male ratio was 1.7:1. A positive family history was documented in seven cases. Three cases developed hypertension and two cases had recurrent urinary tract infections (UTI).
All parents were asymptomatic. Renal ultrasound and urine amino acids were done for the parents and the results were negative.
Biochemical and molecular features (Table 2)
All cases had high urinary dibasic amino acids levels at diagnosis with variable severity. Stone analysis showed yellow–white formation with waxy lusters and cystine. The renal profile remained within normal limits for all patients.
Molecular study showed previously unreported missense variant in exon 10 of SLC3A1 gene (c.1711 T > A p.Cys571Ser) in two cases (Patients 2 and 3). This variant is located in a highly conserved nucleotide and amino acid position with moderate physiochemical differences between the amino acid cysteine and serine. Software analyses by Polyphen-2 and Mutation Taster predicted that the mutation is probably damaging. Another new variant in exon 11 of the SLC9A7 gene (c.1166 C > T p.Thr389Met) was found in one patient (Patient 5). This variant was also located in a highly conserved nucleotide and moderately conserved amino acid position, and there were moderate physiochemical differences between the amino acids threonine and methionine. Software analyses by Polyphen-2 and SIFT predict this variant is probably damaging. A previously reported mutation in exon 8 of the SLC3A1 gene (c.1400 T > A p.Met467Lys), described as disease-causing by Calonge in 1994 (PMID 8054986), was observed in three patients.
Imaging features (Table 3)
All discovered stones were radio-opaque. According to abdominal X-ray, renal ultrasound, and abdominal CT scan studies, we found six cases with multiple bilateral cystine calculi of variable sizes; two of them were staghorn and two cases had unilateral, multiple stones. DMSA scanning showed a unilateral decrease in renal function that had mild severity in two cases and an atrophic small right kidney in another two cases.
Course of the disease
Currently, all patients are in stable condition with variable responses to treatment; five patients were on regular treatment with Tiopronin and potassium citrate and three patients were on penicillamine and potassium citrate. Only one male patient underwent right nephrectomy. Six patients were surgically treated by percutaneous nephrolithotomy.
Discussion
Cystinuria is a rare genetic disease caused by a proximal renal reabsorptive defect of filtered cystine leading to recurrent stone formation [14]. In our study, the age of onset was variable, ranging from birth to 14 years (mean 6.3 years), which is partially consistent with the expected age of the onset of stone formation in cystinuria in the first decade of life and continues lifelong, peaking during the third decade [8, 15]. In a study on Sudanese patients, the mean age at presentation with cystine stones was 31.9 months (range 3–125 months) [16].
In cystinuria, the male to female ratio used to be nearly equal [9]. On the other hand, in our study, females were affected nearly twice as much as males (ratio1.7: 1). Other studies in the Middle East reported a reverse finding with a male to female ratio of 2.3:1 [16]. Two of our patients developed staghorn calculi, supporting the expected natural course of the disease, and they had recurrent and multiple bilateral renal stones [15]. The structure and composition of the stones of our patients mimic the typical composition of cystine stones [16].
Furthermore, 34% of patients with cystine calculi might present with signs of UTI and hypertension; there can be renal failure in up to 17% [6, 7]. In our study, two patients (25%) presented with renal calculi and UTI. Hypertension developed in three cases (37.5%), but there was no renal impairment, which might be due to early detection and proper management.
Although the most frequent mutation in the Mediterranean region in the SLC3A1 gene is M467T [17], three of our patients (37.5%) from a single family were homozygous for the less common M467L mutation, which was previously reported by Calonge, M. et al. (1995) as compound heterozygous. Different regions of the world show characteristic mutation frequencies; for example, p.T216M in SLC3A1 and p.G105R in SLC7A9 are common in Greece [18]. In Portuguese, deletion and duplication (accounts 33.3%), especially E5_E9 Dup, are the most common, while c.1400 T > C p.M467T accounted for 11.1% of mutated alleles. This reflects the importance of screening for large deletion/duplication mutations in cystinuria patients. Furthermore, even if patients were negative, the negative finding does not rule out the disease [10]. Two of our patients (25%) did not show any pathogenic mutations in the target genes; moreover, their deletion/duplication study was also negative. As a result, the patients were diagnosed based on clinical and biochemical results.
In our study, all detected variants were caused by point mutations of the missense type. This is in contrast with previously reported mutations that are nonsense, splicing, small rearrangement and gross rearrangement mutations (insertion and deletion) [10]. Thus far, more than 160 mutations have been discovered in the SLC3A1 gene and more than 116 mutations have been discovered in the SLC7A9 gene. [1].
We identified two novel mutations in the SLC3A1 and SLC7A9 genes. Both variants where located in highly conserved positions, and they are predicted to be damaging according to software analyses. Additionally, they segregated well in the families, which supports the causality for the patient’s phenotype. These newly described variants support and expand on the allelic heterogeneity and complexity of genes variants previously described in cystinuria [19]. The phenotypes of the newly discovered mutations mimic the classical cystinuria phenotype.
Although the missense variant, G105R, has been reported as one of the most frequent mutations in the SLC7A9 gene in cystinuria patients, mainly in Europe [9], it was not found in any of our patients.
In our study, the long-term complications of cystinuria were less than those mentioned in the literature. This may be explained by the early detection and aggressive management of disease. Accordingly, we recommend early screening and diagnosis for other children from affected families.
Conclusion
The newly described mutations result in a classical cystinuria picture. The most frequent mutation discovered in our study was in the SLC3A1 gene, M467L. Negative molecular testing does not rule out the disease if patients are symptomatic. Cystinuria should be considered in each child who has a renal stone and UTI with or without positive family history. Some patients might develop hypertension and UTI, but they do not necessarily have end-stage renal disease especially if diagnosed early and managed properly. No significant clinical differences were detected in this study between types A and B cystinuria. Screening for children in affected families is recommended. A larger data registry is essential for better describing the cystinuria genotype/phenotype in Saudi Arabia.
Abbreviations
- CT:
-
Computed topography
- DMSA:
-
Dimercaptosuccinic acid
- PCR:
-
Polymerase chain reaction
- UTI:
-
Urinary tract infection
References
Andreassen KH, Pedersen KV, Osther SS, Jung HU, Lildal SK, Osther PJ. How should patients with cystine stone disease be evaluated and treated in the twenty-first century? Urolithiasis. 2016;44(1):65–76.
Knoll T, Zollner A, Wendt-Nordahl G, Michel MS, Alken P. Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr Nephrol. 2005;20(1):19–24.
Gitomer WL, Reed BY, Ruml LA, Skhaee K, Pak CYC. Mutations in the genomic deoxyribonucleic acid for SLC3A1 in patients with cystinuria. J Clin Endocrinol Metab. 1998;83(10):3688–94.
Alfadhel M, Benmeakel M, Hossain MA, Al Mutairi F, Al Othaim A, Alfares AA, Al Balwi M, Alzaben A, Eyaid W. Thirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia. Orphanet J Rare Dis. 2016;11(1):126.
Eggermann T, Venghaus A, Zerres K: Cystinuria: an inborn cause of urolithiasis. Orphanet Journal of Rare Diseases 2012, 7(19).
Feliubadaló L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, Golomb E, Centola M, Aksentijevich I, Kreiss Y, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet. 1999;23(1):52–7.
Calonge MJ, Gasparini P, Chillarón J, Chillón M, Gallucci M, Rousaud F, Zelante L, Testar X, Dallapiccola B, Silverio FD, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet. 1994;6:420–5.
Strologo LD. Comparison between SLC3A1 and slc7a9 cystinuria patients and carriers: a need for a new classification. J Am Soc Nephrol. 2002;13(10):2547–53.
Koulivand L, Mohammadi M, Ezatpour B, Salehi R, Markazi S, Dashti S, Kheirollahi M. Mutation analysis of SLC3A1 and SLC7A9 genes in patients with cystinuria. Urolithiasis. 2015;43(5):447–53.
Barbosa M, Lopes A, Mota C, Martins E, Oliveira J, Alves S, De Bonis P, Mota Mdo C, Dias C, Rodrigues-Santos P, et al. Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients. Clin Genet. 2012;81(1):47–55.
Pras E. Cystinuria at the turn of the millennium: clinical aspects and new molecular developments. Mol Urol. 2000;4(4):409–14.
Pierides AM, Deltas CC. Clinical aspects of cystinuria. Hereditary Kidney Dis. 1997;122:167–72.
Dahlberg P, Berg V, Kurtz S, Wilson D, Smith L. Clinical features and management of cystinuria. Mayo Clin Proc. 1977;52(9):533–42.
Al-Marhoon MS, Bayoumi R, Al-Farsi Y, Al-Hinai A, Al-Maskary S, Venkiteswaran K, Al-Busaidi Q, Mathew J, Rhman K, Sharif O, et al. Urinary stone composition in Oman: with high incidence of cystinuria. Urolithiasis. 2015;43(3):207–11.
Al-Hermi B, Abbas B. Cystinuria in Arab Countries. Saudi J Kidney Dis Transplant. 2003;14(1):358–66.
Elfadil GA, Ibrahim ME, Elmugadam AA, Ahmed SAM. Urinary cystine calculi and detection of polymorphism in the SLC3A1 gene in Sudanese children. J Taibah Univ Med Sci. 2014;9(3):219–23.
Botzenhart E, Vester U, Schmidt C, Hesse A, Halber M, Wagner C, Lang F, Hoyer P, Zerres K, Eggermann T, et al. Cystinuria in children: distribution and frequencies of mutations in the SLC3A1 and SLC7A9 genes. Kidney Int. 2002;62(4):1136–42.
Chatzikyriakidou A, Louizou E, Dedousis GV, Bisceglia L, Michelakakis H, Georgiou I. An overview of SLC3A1 and SLC7A9 mutations in Greek cystinuria patients. Mol Genet Metab. 2008;95(3):192–3.
Brons AK, Henthorn PS, Raj K, Fitzgerald CA, Liu J, Sewell AC, Giger U. SLC3A1 and SLC7A9 mutations in autosomal recessive or dominant canine cystinuria: a new classification system. J Vet Intern Med. 2013;27:1400–8.
Acknowledgments
This study could not have been finished without the support and unlimited assistance of the patients and their families.
Funding
This study did not receive fund from any funding body.
Availability of data and materials
The datasets collected and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors’ contributions
AO Performed data collection and analysis and wrote the majority of the manuscript. MN Performed data analysis, wrote and revised the manuscript and submitted the article. KA and AA Provided medical care for the patients, provided patient data, participated in scientific intellectual discussion, and revised and approved of the manuscript. MA Formulated the study design, provided medical care for patients, provided patient data, helped write the manuscript and revised and approved of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
A written consent for publication for this study was obtained from each of the four patients above 16 years and from the legal guardian for each one of the four patients under 16 years included in the study. The written consent included all the data for publication presented in this report. A copy of the written consent is available for review by the editor of this journal.
Ethics approval and consent to participate
The study was approved by the ethics committee at King Abdullah International Medical Research Centre (RC/16/113/R). Inform consent, which includes the use of patients’ data in research, was obtained from patients above 16 years and from legal guardian for patients under 16 years.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Obaid, A., Nashabat, M., Al Fakeeh, K. et al. Delineation of cystinuria in Saudi Arabia: A case series. BMC Nephrol 18, 50 (2017). https://doi.org/10.1186/s12882-017-0469-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-017-0469-x