
Abstract
Background
Joubert syndrome (OMIM 213300) is an autosomal recessive disorder with gene heterogeneity. Causal genes and their variants have been identified by sequencing or other technologies for Joubert syndrome subtypes.
Case presentation
A two-year-old boy was diagnosed with Joubert syndrome by global development delay and molar tooth sign of mid-brain. Whole exome sequencing was performed to detect the causative gene variants in this individual, and the candidate pathogenic variants were verified by Sanger sequencing. We identified two pathogenic variants (NM_006346.2: c.1147delC and c.1054A > G) of PIBF1 in this Joubert syndrome individual, which is consistent with the mode of autosomal recessive inheritance.
Conclusion
In this study, we identified two novel pathogenic variants in PIBF1 in a Joubert syndrome individual using whole exome sequencing, thereby expanding the PIBF1 pathogenic variant spectrum of Joubert syndrome.
Similar content being viewed by others
Background
Joubert syndrome (OMIM: 213300) is an autosomal recessive disorder characterized by a specific mid-hindbrain malformation, hypotonia and developmental delay/intellectual impairment [1]. Molar tooth sign of mid-brain is a diagnostic standard for Joubert syndrome [2]. As Joubert syndrome is a genetically heterogeneous disease, causal genes and their variants have been identified with improved sequencing technologies. Individuals with Joubert syndrome have many of the clinical features of ciliopathies [3], and many ciliary/basal body genes and variants have been discovered to be associated with Joubert syndrome [4, 5]. Joubert syndrome 33 (JBTS33) is caused by PIBF1. Wheway et al. first identified 4 variants or deletions in the PIBF1 gene associated with Joubert syndrome from Hutterite families and other families [6]. A homozygous 36-bp insertion in PIBF1 (c.1181_1182ins36) in a Joubert syndrome family has been reported by Hebbar M. et al. [7]. Moreover, Ott T. et al. found a compound heterozygote (c.1453C > T and c.1508A > G) in a German patient [8].
In this study, we identified two novel pathogenic variants on PIBF1 in a Joubert syndrome individual using whole exome sequencing.
Case presentation
A two-year-old boy was born to a nonconsanguineous couple by cesarean section with a birth weight of 3.42 kg, and the boy’s global development was delayed. He raised his head at 4 months, turned over at 8 months, sat all by himself at 10 months, crawled at 24 months, and stood with support at 28 months. The boy spoke with only monosyllables but had no other verbal communication skills. He had moderate intellectual disability evaluated by the Chinese Developmental Scale for children aged 0–6 years (WS/T 580–2017). The boy had a height of 95 cm (+ 2 SD), weight of 12.4 kg (0 SD), and occipitofrontal circumference of 53 cm (+ 3 SD) at 2 years. Physical examination showed frontal prominence, right eye esotropia, hypotonia and lower myodynamia. Ultrasonic examination showed normal liver, gallbladder, spleen, kidneys, ureter and bladder. Magnetic resonance imaging of his brain revealed unclear vermis of cerebellum, superior cerebellar peduncle thickening and lengthening (a clear molar tooth sign) as well as cerebellar hemispheres joining in the midline of brain and a smaller midbrain (Fig. 1). Thus, the boy was diagnosed with Joubert syndrome.

Brain magnetic resonance imaging (MRI) findings for the Joubert syndrome individual. Molar tooth sign with moderate cerebellar vermis hyoplasia, lengthening and thickening of superior cerebellar peduncles as well as superior cerebellar dysplasia as indicated by white arrows. a T2-weighted image. b T1-weighted image. c Pedigree of Joubert syndrome patient
EDTA anticoagulant venous blood samples (2 mL) were obtained from all affected and unaffected family members. Genomic DNA was extracted from whole blood using the QIAamp® DNA Blood Mini Kit (QIAGEN, Germany) according to the manufacturer’s protocol, and whole exome sequencing was performed for the patient. An Agilent SureSelect Human All Exon V6 kit (Agilent Technologies Inc., USA) was used for preparation of the exome library using appromximately 3 μg of genomic DNA. The exome library was sequenced with a mean 100× coverage on an Illumina NovaSeq 6000 platform (Illumina Inc., USA). The average target coverage depth was 126× with > 97% of the bases covered at >20x, and the sensitivity was > 98%.
Raw data that passed quality control was aligned to the human reference genome (GRCh37/hg19), and only high-quality data (>Q30) was used for variant calling. Alignment to the human reference genome (GRCh37/hg19) and variant calling were performed using Burrows-Wheeler Aligner software (http://bio-bwa.sourceforge.net), followed by variant annotation by ANNOVAR [9] with the dbSNP147 databases (https://www.ncbi.nlm.nih.gov/SNP/), 1000G database (http://www.1000genomes.org/), ExAC database (http://exac.hms.harvard.edu/), HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) and OMIM (https://www.ncbi.nlm.nih.gov/omim/).
The filtering criteria are shown in supplementary Table 1. The filtered variants were classified by the American College of Medical Genetics and Genomics (ACMG) standard to determine the pathogenic genes and variant sites. Conservation of different species was analyzed by MEGA6.0 software [10]. The candidate variants identified via whole exome sequencing were validated, and segregation analysis was performed by Sanger sequencing using the ABI3730xl Genetic Analyzer (Life Technologies, Carlsbad, CA) following the manufacturer’s protocol. The Sanger sequencing chromatogram was viewed by Chromas software and aligned to other reference sequences by SeqMan software. The PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), PROVEAN (http://provean.jcvi.org/index.php) and Mutation Taster (http://www.mutationtaster.org/) online tools were used to predict the effect of candidate variants for protein function. The protein domains were analyzed by the SMART online service (http://smart.embl-heidelberg.de/).
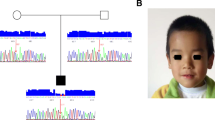
Whole exome sequencing identified one variant (c.1626 + 1G > A) in AHI1 (NM_001134832.1) and two variants (c.1147delC and c.1054A > G) in PIBF1 (NM_006346.2) in a heterozygous state in the affected individual. Direct Sanger sequencing of the patient and parents showed that AHI1 c.1626 + 1G > A was from his father (heterozygote) as his mother was wild-type for this variant. For PIBF1, only one heterozygote of these two variants was observed in his parents (c.1147delC in mother and c.1054A > G in father) (Fig. 2).

Sanger chromatograms. a c.1147delC variant in PIBF1 in a heterozygous state in the proband (upper panel) and a heterozygous state in the mother (middle panel) and a wild-type homozygous state in the father (lower panel). b c.1054A > G variant in PIBF1 in a heterozygous state in the proband (upper panel) and a wild-type homozygous state in the mother (middle panel) and a heterozygous state in the father (lower panel). c c.1626 + 1G > A variant in AHI1 in a heterozygous state in the proband (upper panel) and a wild-type homozygous state in the mother (middle panel) and a heterozygous state in the father (lower panel)
These two PIBFI variants were absent in the 1000 Genomes Project, ESP, gnomAD, Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC) datasets. The c.1147delC variant is located in exon 9 of PIBF1, leading to an amino acid change from glutamine to lysine at the position 383 of the PIBF1 protein, which causes a frameshift, resulting a premature protein truncation (p.Gln383LysfsTer4). The c.1054A > G variant is located in exon 8 of PIBF1, and it is a missense variant in exon 8, leading to an amino acid change from lysine to glutamic acid at position 352 of the PIBF1 protein (p.Lys352Glu). This variant was predicted to be probably damaging, deleterious and disease causing according to the protein predicted by PolyPhen-2 with a score of 0.962, PROVEAN with a score of − 2.833, and Mutation Taster with a score of > 0.999, respectively. According to the ACMG standards and guidelines, these PIBF1 variants were classified as pathogenic (c.1147delC) and likely pathogenic (c.1054A > G) [11].
Discussion and conclusions
We identified two novel pathogenic variants (NM_006346.2: c.1147delC and c.1054A > G) in PIBF1 by whole exome sequencing of a Joubert syndrome individual. These two variants were present in heterozygous state in the affected child, which was consistent with the autosomal recessive inheritance mode. The c.1147delC variant was maternally inherited, and the c.1054A > G variant was paternally inherited.
PIBF1 is located in chromosome 13q21-q22, contains 18 exons and spans more than 234 kb [12], and it encodes a predicted hydrophilic 757-amino acid alpha-helical protein [13], which is produced during pregnancy in response to progesterone [14]. Kim K. et al. reported that the PIBF1 protein plays an important role in the formation of primary cilia [15]. PIBF1 is a core component of the human centrosome and is crucial for the accumulation of centriolar satellites, eventually forming the primary cilia [15]. Depletion of PIBF1 causes mitotic arrest, misaligned chromosomes and spindle pole fragmentation [15]. Exogenous expression of human wild-type PIBF1 following siRNA knockdown rescues ciliogenesis in mIMCD3 cells [6]. A whole genome siRNA reverse genetics screen has identified recessive variants in PIBF1 in seven individuals with Joubert syndrome [6].
Some variants of PIBF1 have been identified to be associated with Joubert syndrome (Table 1) and all the variants are showed in Fig. 3. In this study, we found two novel variants in a Joubert syndrome individual. All Joubert syndrome individuals with PIBF1 variants have a developmental delay and hypotonia, and most of these individuals have molar tooth sign and cerebellar vermis hypoplasia. Abnomal ocular movement was present in the affected boy of the present study and in a girl previously reported by Ott. T et al. Only Hutterite descent individuals shown the syndrome of ataxia (Table 1). Due to limited cases, the relationship of variant type and Joubert syndrome symptoms needs further investigation.

Pathogenic variants in the PIBF1 gene and the protein structure. The novel variants of this study are marked in red. CC, coiled coil region; lc, low complexity region
A reported pathogenic variant (NM_001134832.1 c.1626 + 1G > A) in the AHI1 gene was found in a heterozygous state in the patient and his father but as a wild-type in his mother, indicating that this variant did not segregate in the family. AHI1 is located in chromosome 6q23.3, contains 33 exons and spans more than 213 kb [16]. Ferland R.J. et al. first reported the relationship between Joubert syndrome and AHI1 variants [17]. Parisi, M. A. et al. reported that variants of AHI1 cause both retinal dystrophy and renal cystic disease in Joubert syndrome patients [18]. To date, more than 10 variants associated with Joubert syndrome have been reported [17, 19,20,21,22,23]. AHI1 c.1626 + 1G > A represents a G to A transition at the first base downstream of the 3′ end of exon 12 in AHI1 as a splice variant. This variant was reported by Bachmann-Gagescu, R. et al., as a compound heterozygote combined with the c.2361G > T variant in AHI1 to cause Joubert syndrome [19]. We screened all of the AHI1 exons by Sanger sequencing for this Joubert syndrome patient and his parents, and we did not find another pathogenic or likely pathogenic variant in AHI1, except for c.1626 + 1G > A. Therefore, this variant was not the pathogenic cause for this child (data not shown).
Next generation sequencing has aided in the search for genetic variants of rare inherited diseases, such as Joubert syndrome. To date, however, Joubert syndrome can be treated but not cured. Thus, it is important to understand the underlying disease mechanism. Additional functional information is required to develop a treatment and comprehend the developmental regularity of the inherited diseases.
In conclusion, the present study identified two novel variants in PIBF1 associated with Joubert syndrome and expanded the PIBF1 pathogenic variant spectrum of Joubert syndrome. Further functional validation is necessary to clarify the pathogenic mechanism of the PIBF1 gene in Joubert syndrome.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the human reference 8 genome (GRCh37/hg19), NM_001134832.1, NM_006346.2.
Abbreviations
- PIBF1 :
-
Progesterone immunomodulatory binding factor 1
- JBTS:
-
Joubert syndrome
References
Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007;15(5):511–21.
Nag C, Ghosh M, Das K, Ghosh T. Joubert syndrome: the molar tooth sign of the mid-brain. Ann Med Health Sci Res. 2013;3(2):291–4.
Sattar S, Gleeson JG. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol. 2011;53(9):793–8.
Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009;151C(4):326–40.
Davis EE, Katsanis N. The ciliopathies: a transitional model into systems biology of human genetic disease. Curr Opin Genet Dev. 2012;22(3):290–303.
Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen TT, Racher H, Phelps IG, Toedt G, Kennedy J, Wunderlich KA, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol. 2015;17(8):1074–87.
Hebbar M, Kanthi A, Shukla A, Bielas S, Girisha KM. A biallelic 36-bp insertion in PIBF1 is associated with Joubert syndrome. J Hum Genet. 2018;63(8):935–9.
Ott T, Kaufmann L, Granzow M, Hinderhofer K, Bartram CR, Theiss S, Seitz A, Paramasivam N, Schulz A, Moog U, et al. The frog Xenopus as a model to study Joubert syndrome: the case of a human patient with compound heterozygous variants in PIBF1. Front Physiol. 2019;10:134.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Rozenblum E, Vahteristo P, Sandberg T, Bergthorsson JT, Syrjakoski K, Weaver D, Haraldsson K, Johannsdottir HK, Vehmanen P, Nigam S, et al. A genomic map of a 6-Mb region at 13q21-q22 implicated in cancer development: identification and characterization of candidate genes. Hum Genet. 2002;110(2):111–21.
Polgar B, Kispal G, Lachmann M, Paar C, Nagy E, Csere P, Miko E, Szereday L, Varga P, Szekeres-Bartho J. Molecular cloning and immunologic characterization of a novel cDNA coding for progesterone-induced blocking factor. J Immunol. 2003;171(11):5956–63.
Szekeres-Bartho J, Kilar F, Falkay G, Csernus V, Torok A, Pacsa AS. The mechanism of the inhibitory effect of progesterone on lymphocyte cytotoxicity: I. progesterone-treated lymphocytes release a substance inhibiting cytotoxicity and prostaglandin synthesis. Am J Reprod Immunol Microbiol. 1985;9(1):15–8.
Kim K, Lee K, Rhee K. CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS One. 2012;7(10):e48196.
Jiang X, Hanna Z, Kaouass M, Girard L, Jolicoeur P. Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J Virol. 2002;76(18):9046–59.
Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36(9):1008–13.
Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43(4):334–9.
Bachmann-Gagescu R, Dempsey JC, Phelps IG, O'Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52(8):514–22.
Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, Barrano G, Bertini E, Boltshauser E, Zaki MS, Abdel-Aleem A, et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol. 2006;59(3):527–34.
Elsayed SM, Phillips JB, Heller R, Thoenes M, Elsobky E, Nurnberg G, Nurnberg P, Seland S, Ebermann I, Altmuller J, et al. Non-manifesting AHI1 truncations indicate localized loss-of-function tolerance in a severe Mendelian disease gene. Hum Mol Genet. 2015;24(9):2594–603.
Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, Hennies HC, Otto EA, Hildebrandt F. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21(1):32–5.
Tuz K, Hsiao YC, Juarez O, Shi B, Harmon EY, Phelps IG, Lennartz MR, Glass IA, Doherty D, Ferland RJ. The Joubert syndrome-associated missense mutation (V443D) in the Abelson-helper integration site 1 (AHI1) protein alters its localization and protein-protein interactions. J Biol Chem. 2013;288(19):13676–94.
Acknowledgements
We are thankful for participation of the patient family in this study.
Funding
The work was supported by the following: the National Key Research and Development Program of China (2016YFC1000307) to Xu Ma; and the Non-profit Central Research Institute Fund of National Research Institute for Family Planning (2018GJZ02 and 2020GJZ05) to Minna Luo. The funders had no role in study design, data collection and analysis as well as decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YS, HW and ZL were the major contributors in writing the manuscript. XM and BX conceived the concept and supervised the studies. HW and ZL collected the blood samples. HW, ZL, YP and BX collected and interpreted clinical data of the patients. YS, ML, QL, SM and CL contributed to DNA extraction and sequencing experiments. ZC, YY, RC, CC and HG analyzed the sequencing data. All authors read and approved the submitted version of manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all participants (the patient’s consent was signed by the parents). This study was approved by the Ethical Committees of the National Research Institute for Family Planning, and it clearly stated that blood samples would be used for scientific research purposes, including genetic studies.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, Y., Wang, H., Liu, Z. et al. Identification of two novel pathogenic variants of PIBF1 by whole exome sequencing in a 2-year-old boy with Joubert syndrome. BMC Med Genet 21, 192 (2020). https://doi.org/10.1186/s12881-020-01130-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01130-x