
Abstract
Background
Amelogenesis imperfecta (AI) is a highly heterogeneous group of hereditary developmental abnormalities which mainly affects the dental enamel during tooth development in terms of its thickness, structure, and composition. It appears both in syndromic as well as non-syndromic forms. In the affected individuals, the enamel is usually thin, soft, rough, brittle, pitted, chipped, and abraded, having reduced functional ability and aesthetics. It leads to severe complications in the patient, like early tooth loss, severe discomfort, pain, dental caries, chewing difficulties, and discoloration of teeth from yellow to yellowish-brown or creamy type. The study aimed to identify the disease-causing variant in a consanguineous family.
Methods
We recruited a consanguineous Pashtun family of Pakistani origin. Exome sequencing analysis was followed by Sanger sequencing to identify the pathogenic variant in this family.
Results
Clinical analysis revealed hypomaturation AI having generalized yellow-brown or creamy type of discoloration in affected members. We identified a novel nonsense sequence variant c.1192C > T (p.Gln398*) in exon-12 of SLC24A4 by using exome sequencing. Later, its co-segregation within the family was confirmed by Sanger sequencing. The human gene mutation database (HGMD, 2019) has a record of five pathogenic variants in SLC24A4, causing AI phenotype.
Conclusion
This nonsense sequence variant c.1192C > T (p.Gln398*) is the sixth disease-causing variant in SLC24A4, which extends its mutation spectrum and confirms the role of this gene in the morphogenesis of human tooth enamel. The identified variant highlights the critical role of SLC24A4 in causing a rare AI type in humans.
Similar content being viewed by others

Background
Mature enamel is a thin outer protective layer and covers the crown of the tooth in the form of a shell [1]. Naturally, it is tough, hard, and highly mineralized translucent human tissue produced by ameloblasts and is epithelial in its origin [2]. The biochemical architecture of dental enamel is of crystals of substituted calcium hydroxyapatite (96%), and the 4% is of organic matter and water [3]. Amelogenesis is a highly intricate biomineralizing process controlled by the expression of several genes [2]. AI affects both the primary and permanent dentition with exceptionally variable severity of the disease conditions [4, 5].
Various accounts of both syndromic and non-syndromic/isolated cases of AI have been published in the literature. Depending upon the amount, structure, and composition of the dental enamel, the phenotypes of non-syndromic AI are highly variable and may be divided into hypoplastic, hypocalcified, and hypomaturation forms [3, 4].
To date, pathogenic variants causing non-syndromic AI have been identified in 20 genes at various chromosomal locations [3], including AMELX (OMIM 300391; Xp22.2), a candidate gene for X-linked dominant hypoplastic AI (OMIM: 301200) [6], encoding an enamel matrix protein (EMPs) called amelogenin and making up to 90% of the ameloblast secreted EMPs [7, 8]. ENAM (OMIM 606585; 4q13.3), encoding the largest EMP called enamelin, a tooth specific protein expressed by ameloblasts, causing an autosomal recessive (OMIM: 204650) and dominant forms of AI (OMIM 104500) [9, 10]. AMBN (OMIM 601259; 4q13.3) encodes a glycine, leucine, and proline-rich enamel matrix protein called ameloblastin, a second most abundant protein expressed during amelogenesis. AMBN associated AI segregates in an autosomal recessive fashion [11, 12]. LAMB3 (OMIM 150310; 1q32.2), LAMA3 (OMIM 600805; 18q11.2), COL17A1 (OMIM 113811; 10q25.1), ITGB6 (OMIM 147558; 2q24.2) and ACPT (OMIM: 606362; 19q13.33) are other genes that cause hypoplastic AI in their altered forms [13,14,15,16,17,18,19,20]. Mutations in FAM83H (OMIM 611927; 8q24.3) cause an autosomal dominant hypocalcified type of AI [6, 21]. However, SLC24A4 (OMIM 609840; 14q32.12), WDR72 (OMIM 613214; 15q21.3), MMP20 (OMIM 604629; 11q22.2), KLK4 (OMIM 603767; 19q13.41) and GPR68 (OMIM 601404; 14q32.11), cause autosomal recessive hypomaturation type of AI [6, 22,23,24,25,26]. MMP20 (OMIM 604629; 11q22.2) and KLK4 (OMIM 603767; 19q13.41) are the two proteinases secreted at the time of enamel formation [27]. Nevertheless, in the case of C4orf26 (OMIM 614829; 4q21.1), and AMTN (OMIM 610912; 4q13.3) mutations cause autosomal recessive and dominant hypo-mineralized amelogenesis imperfecta, respectively [28, 29]. Recently, RELT (OMIM 611211; 11q13.4) variants are identified, causing hypocalcified amelogenesis imperfecta type IIIC [30].
Occasionally, AI has been reported as a part of a syndrome. The most common of them include Tricho-Dento-Osseous (TDO; OMIM 190320) syndrome (DLX3, OMIM 600525), Laryngo-Onycho-Cutaneous (LOC; OMIM 245660) syndrome (LAMA3, OMIM 600805), Jalili syndrome (JS; OMIM 217080) (CNNM4, OMIM 607805), Amelogenesis Imperfecta and Nephrocalcinosis (OMIM 204690) (FAM20A, OMIM 611062), Kohlschutter-Tonz Syndrome (KTS; MIM 226750) (ROGDI, OMIM 614574), Amelo-Onycho-Hypohidrotic Syndrome (OMIM 104570), and Heimler Syndrome-1,2 (HMLR; OMIM 234580) (PEX1, PEX6, OMIM 602136, 601,498).
Here, we report a novel nonsense variant c.1192C > T (p.Gln398*) in exon-12 of SLC24A4 in non-syndromic AI patients in a family of Pakistani origin.
Methods
Patients recruitment, pedigree construction, and DNA extraction
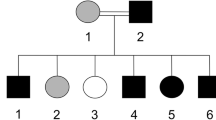
The recommendations of the Declarations of Helsinki were strictly followed for the approval of the study from the Research and Ethical Committee of Kohat University of Science and Technology (KUST), Khyber Pakhtunkhwa, Pakistan. Informed written consent was obtained from the affected and unaffected participants. A five generational pedigree diagram was constructed after a thorough interview of the unaffected mother (III-4). The pedigree showed an autosomal recessive mode of inheritance (Fig. 1A). Venous blood samples were collected from seven members of the family, including two patients (IV-4, IV-5) and five phenotypically unaffected individuals (III-4, IV-1, IV-3, IV-7, V-1). The extraction of genomic DNA from whole peripheral blood was performed by using the GeneJET Genomic DNA extraction Kit (Thermo-scientific, Lithuania), strictly following the manufacturer’s protocol.

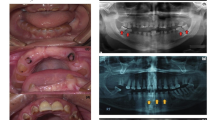
(A) Pedigree of the family where SLC24A4 variant c.1192C > T segregates in an autosomal recessive fashion. The asterisks show tested individuals. The red arrow indicates the index patient, who was subjected toexome sequencing. T shows the disease-allele while C is the wild-type presentation (B) The representation of amelogenesis imperfecta in the patients. (i), (ii), and (iii) are the clinical features of patient IV-4 showing yellow-brown discoloration, (iv) Orthopantomogram (OPG) of the patient IV-4 showing thin layer of enamel, high radio-density and distinction from the dentin, while (v) and (vi) are the clinical photographs of the patient IV-5 showing creamy type of discoloration, attrition and dental caries
Exome sequencing, and validation of rare variants through DNA sequencing
DNA (70 ng/μl) of an affected member (IV-4) was subjected to exome sequencing. The enrichment of DNA for the intron-exon boundaries was carried out with the SeqCap EZ human exome library v2.0 kit. The Illumina HiSeq 4000 sequencing machine via a paired-end 100-bp protocol [31] was used to run the generated libraries. The Cologne Center for Genomics (CCG) Varbank pipeline v2.26 (https://varbank.ccg.uni-koeln.de/) was used for exome data analysis. The mean coverage of the data was 77%, while at 20X and 10X, the coverage of the targeted bases was 92.6 and 96.6%, respectively. Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/) was consulted to establish the minor allele frequency (MAF; value < 0.01) of the variants. As controls, an in-house database of 511 exomes, and a dataset of 65 exomes from the Pakistani population, including 44 exomes from Punjabi, Sindhi, and Balochi patients, and 21 exomes of ethnically matched Pakhtoon patients were also consulted. The rare variants in PSPH, CHCHD2, BNC2, and SLC24A4 were selected from the exome data and were considered for the co-segregation analysis. The online prediction tools like MutationTaster, PROVEAN, SIFT, and PolyPhen2.0 were used to predict the pathogenicity of the missense variants (Table 1). The reference sequences of PSPH, CHCHD2, BNC2, and SLC24A4 (NM_004577.3, NM_016139.2, NM_017637.5, NM_153646.3, respectively) were obtained from the University of California Santa Cruz (UCSC) genome database browser (http://genome.ucsc.edu/cgi-bin/hgGateway). Primer3Plus software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) was used for designing the primers for the amplification of the regions of interest. A nucleotide sequence of 600 bp up-and-downstream from the position of the rare variants was searched to find out a suitable primer pair (Table 1). PCR amplified the regions of interest and the Exo-Sap protocol (https://www.thermofisher.com) was used for purifying the PCR products. The DNA sequencing was performed on the ABI3730 genetic analyzer with BigDye chemistry v3.1. The sequence alignment against the reference sequence was carried out by a sequence alignment tool, BioEdit version 6.0.7 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).
Results
Clinical and radiological investigations
For clinical and radiological investigations, a 35-years old patient (IV-4) was referred to Khyber Medical College of Dentistry, Peshawar, Khyber Pakhtunkhwa, Pakistan. His major complaints were yellow-brown staining, eating, and chewing difficulties of all the teeth (Fig. 1B-i,ii,iii). The patient presented no complications of other body organs during the clinical evaluation. The Orthopantomogramm (OPG) of this patient showed a thin (hypoplastic) mandible with missing posterior teeth on the right side and carious molars with a periapical infection on the left side. The maxilla showed impacted canine in the right premolar region with a missing molar and spacing among the dentition on the right side of the arch.
Additionally, the teeth showed generalized horizontal bone loss, more prominent around the maxillary molars. OPG also showed the presence of a thin layer of enamel, especially in the region of molars of the upper jaw. Furthermore, enamel appeared to have higher radio-density compared to the dentin. Moreover, the dentin appeared normal and distinct from the enamel (Fig. 1B-iv).
Patient IV-5, the 27-year old brother of patient IV-4, presented with creamy discoloration and attrition of the frontal maxillary teeth while dental caries in the mandibular premolars and molars (Fig. 1B-v,vi).
Screening of pathogenic sequence variant
Exome sequencing revealed rare homozygous variants in four genes: PSPH (OMIM 172480; Exon-6, c.398A > G; p.Arg133Ser), CHCHD2 (OMIM 616244; Exon-3, c.418G > A; p.Val140Met), BNC2 (OMIM 608669; Exon-7, c.2860G > A; p.Ala954Thr), and SLC24A4 (c.1192C > T; p.Gln398*). These variants lie in three regions of homozygosity (ROH) on chromosome 7 (11.6 MB), 9 (3.8 MB), and 14 (4.7 MB). The variants in CHCHD2 and SLC24A4 are neither reported in gnomAD nor HGMD (Human Gene Mutation Database; http://www.hgmd.cf.ac.uk/ac/index.php). Both variants in PSPH and BNC2 are tremendously rare in gnomAD, where c.398A > G; p.Arg133Ser appears in 20 alleles out of 282,490 alleles (none homozygous) and c.2860G > A; p.A954T is found in 4 alleles (one is homozygous) out of 246,026 alleles. These variants are not identified in the in-house database of 511 exomes and 65 exomes of Pakistani patients with diverse phenotypes other than AI. The pathogenicity predictions of the variants in PSPH, CHCHD2, and BNC2 by four online prediction algorithms are described in Table 1.
Sanger sequencing was used to check the segregation of these variants with the disease. The homozygous missense variants in PSPH, CHCHD2, BNC2 did not segregate within the family while the homozygous nonsense variant (c.1192C > T; p.Gln398*) in SLC24A4 revealed its co-segregation in the family (Fig. 2A). The DNA sequencing results of this cohort showed three forms of genotypes for this variant, heterozygous (C/T) (III-4, IV-3, IV-7), homozygous (C/C) wild-type (IV-1, V-1) and homozygous (T/T) mutant (IV-4, IV-5) (Fig. 1A). A ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/variation/689492/) accession number (VCV000689492.1) for this variant has been allocated.

a Chromatograms of an unaffected individual (IV-1) in the upper panel, a carrier (III-4) member in the middle panel and an affected individual IV-4 in the lower panel. b Hypothetical structure of SLC24A4 containing all 17 exons, showing the positions of genetic alterations in the previous studies as well as in the present study (red)
Exome data did not expose any rare variant in other genes (AMELX, ENAM, AMBN, LAMB3, LAMA3, COL17A1, ITGB6, ACPT, FAM83H, WDR72, MMP20, KLK4, GPR68, RELT, DLX3, CNNM4, ROGDI, PEX1, and PEX6) reported so far, to cause syndromic and non-syndromic AI.
Discussion
Five functionally different types of K+-dependent Na+/Ca+ 2 exchangers (NCKX1–5) have been characterized in humans [32, 33]. NCKXs are bidirectional membrane transporters; for example, NCKX4 transports an intracellular Ca+ 2 and a K+ ion in exchange for four extracellular Na+ ions [34]. Each NCKX protein has a unique role in various biochemical pathways governing the vision, olfaction, and skin pigmentation [35]. During the maturation stage of tooth development, SLC24A4 (NCKX4) is involved in the active transport of Ca+ 2 ions from ameloblasts into the enamel matrix. Genetic alterations in SLC24A4 in the human genome and its knockout mice Slc24a4−/− lead to the development of indisposed calcified enamel [36]. Clinical findings of Slc24a4−/− mice signify the essential role of this protein in enamel development [25].
SLC24A4 (OMIM 609840) encodes a protein of 622 amino acids, called solute carrier family 24 member 4 (SLC24A4), which is one of the members of K+-dependent Na+/Ca2+ exchanger family (SLC24A), comprising a total of five members. It has been mapped to the chromosome 14q32 [33, 36]. SLC24A4 has various transcripts (NM_153646, NM_153647, NM_153648) resulting from alternative splicing and the longest isoform (NM_153646) contains 17 coding exons. SLC24A4 is highly expressed in many types of tissues, such as aorta, brain, lungs, and thymus gland [34]. In the case of developing dentine, SLC24A4 is expressed in ameloblasts, and it borders to the membrane in contact with the developing enamel [37]. The predicted structure for full-length SLC24A4 protein consists of 11 transmembrane helices having two highly conserved transmembrane clusters (consisting of 5 transmembrane helices) linked together by an intracellular (cytoplasmic) loop. The Na+/Ca2+ exchanger domains are composed of these transmembrane pockets. Each domain contains a hydrophobic and highly conserved region of 30–40 residues called alpha-1 (139–179 amino acids), and alpha-2 (495–526 amino acids) repeats, respectively, which form ion-binding regions after undergoing highly intricate interactions with each other [25, 38].
We have identified a novel nonsense variant (c.1192C > T; p.Gln398*) in exon-12 of SLC24A4 by using exome sequencing. This unusual genetic alteration is expected to lead to the loss of function of SLC24A4 protein either by nonsense-mediated decay (NMD) or by the production of a truncated protein lacking the C-terminus. Since this nonsense variant introduces a premature stop codon at the position 398 in the cytoplasmic loop between the alpha-1 and alpha-2 repeats; hence the loss of remaining 225 amino acids (containing the alpha-2 repeat) is predicted. The two Na+/Ca2+ exchanger domains (alpha-1 and alpha-2 repeats) are crucial for the smooth transport of ions, which verifies the exceptional role of SLC24A4 during amelogenesis. The absence of one of the two Na+/Ca2+ exchanger domains, in this case, alpha-2-repeat only will ultimately render the protein nonfunctional and causes amelogenesis imperfecta, hypomaturation type AI2A5 (OMIM: 615887) phenotype [25].
To date, a total of five pathogenic variants causing AI have been identified in the SLC24A4, including three missense variants, one nonsense variant, and a gross deletion (Fig. 2B). Parry et al. in 2013 screened 15 Pakistani families and identified two homozygous variants in SLC24A4, including a missense c.1495A > T (p.Ser499Cys), and a nonsense variant c.1015C > T (p.Arg339*) in two consanguineous families. They performed Sanger sequencing of 37 AI patients of different ethnicities and suggested that pathogenic sequence variants in SLC24A4 are a rare cause of AI in general, but might be a frequent cause of AI in Pakistani population [25]. Researches on three consanguineous Turkish families have revealed two homozygous missense pathogenic variants c.437C > T; (p.Ala146Val), c.1317 T > G (p.Leu436Arg) and a 10 kb (10,042 bp) homozygous deletion, comprising of exons 15, 16 and most of the exon-17 (Chr14: 92,957,680-92,967,722del) [36, 39, 40]. During a comparison of AI phenotypes caused by SLC24A4 variants in patients reported so far in the literature, we have concluded that clinical manifestation of AI is moderately to severely variable among the cases (Table 2).
Conclusion
The present study aimed to perform a clinical and molecular evaluation of an autosomal recessive Pakistani family. We have identified the sixth disease-causing variant in SLC24A4 (Fig. 2B), which extends its mutation spectrum and confirms the role of this gene in the morphogenesis of human tooth enamel.
Availability of data and materials
The data generated during the current study are available on online public repository ClinVar (https://submit.ncbi.nlm.nih.gov/clinvar/). An accession number (VCV000689492.1) for the novel variant identified in this study has also been allocated (https://www.ncbi.nlm.nih.gov/clinvar/variation/689492/). If any further information is needed, please ask the corresponding authors.
Abbreviations
- AI:
-
Amelogenesis imperfecta
- HGMD:
-
Human Gene Mutation Database
- TDO:
-
Tricho-Dento-Osseous
- LOC:
-
Laryngo-Onycho-Cutaneous
- JS:
-
Jalili syndrome
- KTS:
-
Kohlschutter-Tonz Syndrome
- HMLR:
-
Heimler Syndrome
- KUST:
-
Kohat University of Science and Technology
- CCG:
-
Cologne Center for Genomics
- gnomAD:
-
Genome Aggregation Database
- MAF:
-
Minor allele frequency
- UCSC::
-
University of California Santa Cruz
- OPG:
-
Orthopantomogramm
- NMD:
-
Nonsense-mediated decay
References
Eastoe JE. Organic matrix of tooth enamel. Nature. 1960;187:411–2.
Paine ML, et al. Regulated gene expression dictates enamel structure and tooth function. Matrix Biol. 2001;20(5–6):273–92.
Smith CEL, et al. Amelogenesis Imperfecta; genes, proteins, and pathways. Front Physiol. 2017;8:435.
Witkop CJ Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol. 1988;17(9–10):547–53.
Crawford PJ, Aldred M, Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J Rare Dis. 2007;2:17.
Wright JT, et al. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res. 2003;44(Suppl 1):72–8.
Lagerstrom M, et al. A deletion in the amelogenin gene (AMG) causes X-linked amelogenesis imperfecta (AIH1). Genomics. 1991;10(4):971–5.
Termine JD, et al. Properties of dissociatively extracted fetal tooth matrix proteins. I. Principal molecular species in developing bovine enamel. J Biol Chem. 1980;255(20):9760–8.
Hu JC, Yamakoshi Y. Enamelin and autosomal-dominant amelogenesis imperfecta. Crit Rev Oral Biol Med. 2003;14(6):387–98.
Hart TC, et al. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 2003;40(12):900–6.
MacDougall M, et al. Ameloblastin gene (AMBN) maps within the critical region for autosomal dominant amelogenesis imperfecta at chromosome 4q21. Genomics. 1997;41(1):115–8.
Poulter JA, et al. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum Mol Genet. 2014;23(20):5317–24.
Kim JW, et al. LAMB3 mutations causing autosomal-dominant amelogenesis imperfecta. J Dent Res. 2013;92(10):899–904.
Poulter JA, et al. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet. 2014;22(1):132–5.
Lee KE, et al. Novel LAMB3 mutations cause non-syndromic amelogenesis imperfecta with variable expressivity. Clin Genet. 2015;87(1):90–2.
Yuen WY, et al. Enamel defects in carriers of a novel LAMA3 mutation underlying epidermolysis bullosa. Acta Derm Venereol. 2012;92(6):695–6.
McGrath JA, et al. Compound heterozygosity for a dominant glycine substitution and a recessive internal duplication mutation in the type XVII collagen gene results in junctional epidermolysis bullosa and abnormal dentition. Am J Pathol. 1996;148(6):1787–96.
Wang SK, et al. ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Hum Mol Genet. 2014;23(8):2157–63.
Seymen F, et al. Recessive mutations in ACPT, encoding testicular acid phosphatase, cause Hypoplastic Amelogenesis Imperfecta. Am J Hum Genet. 2016;99(5):1199–205.
Smith CE, et al. Defects in the acid phosphatase ACPT cause recessive hypoplastic amelogenesis imperfecta. Eur J Hum Genet. 2017;25(8):1015–9.
Kim JW, et al. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet. 2008;82(2):489–94.
Hart PS, et al. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004;41(7):545–9.
Kim JW, et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005;42(3):271–5.
El-Sayed W, et al. Mutations in the beta propeller WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet. 2009;85(5):699–705.
Parry DA, et al. Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am J Hum Genet. 2013;92(2):307–12.
Parry DA, et al. Mutations in the pH-sensing G-protein-coupled receptor GPR68 cause Amelogenesis Imperfecta. Am J Hum Genet. 2016;99(4):984–90.
Hu JC, et al. Enamelysin and kallikrein-4 mRNA expression in developing mouse molars. Eur J Oral Sci. 2002;110(4):307–15.
Parry DA, et al. Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. Am J Hum Genet. 2012;91(3):565–71.
Smith CE, et al. Deletion of amelotin exons 3-6 is associated with amelogenesis imperfecta. Hum Mol Genet. 2016;25(16):3578–87.
Kim JW, et al. Mutations in RELT cause autosomal recessive amelogenesis imperfecta. Clin Genet. 2019;95(3):375–83.
Hussain MS, Baig SM, Neumann S, Peche VS, Szczepanski S, Nürnberg G, Tariq M, Jameel M, Khan TN, Fatima A, Malik NA, Ahmad I, Altmüller J, Frommolt P, Thiele H, Höhne W, Yigit G, Wollnik B, Neubauer BA, Nürnberg P, Noegel AA. CDK6 associates with the centrosome during mitosis and is mutated in large Pakistani family with primary microcephaly. Hum Mol Genet. 2013;22(25):5199–214. https://doi.org/10.1093/hmg/ddt374.
Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406(3):365–82.
Szerencsei RT, et al. The Na/Ca-K exchanger gene family. Ann N Y Acad Sci. 2002;976:41–52.
Li XF, Kraev AS, Lytton J. Molecular cloning of a fourth member of the potassium-dependent sodium-calcium exchanger gene family, NCKX4. J Biol Chem. 2002;277(50):48410–7.
Jalloul AH, et al. A functional study of mutations in K+-dependent Na+−Ca2+ exchangers associated with Amelogenesis Imperfecta and non-syndromic Oculocutaneous albinism. J Biol Chem. 2016;291(25):13113–23.
Wang S, et al. STIM1 and SLC24A4 Are Critical for Enamel Maturation. J Dent Res. 2014;93(7 Suppl):94s–100s.
Hu P, et al. Expression of the sodium/calcium/potassium exchanger, NCKX4, in ameloblasts. Cells Tissues Organs. 2012;196(6):501–9.
Iwamoto T, et al. The Na+/Ca2+ exchanger NCX1 has oppositely oriented reentrant loop domains that contain conserved aspartic acids whose mutation alters its apparent Ca2+ affinity. J Biol Chem. 2000;275(49):38571–80.
Seymen F, et al. Exonal deletion of SLC24A4 causes hypomaturation amelogenesis imperfecta. J Dent Res. 2014;93(4):366–70.
Herzog CR, et al. Hypomaturation amelogenesis imperfecta caused by a novel SLC24A4 mutation. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;119(2):e77–81.
Acknowledgments
We are thankful to family members for their total contribution. We sincerely acknowledge the services of the medical staff at Khyber Medical College of Dentistry, Peshawar, Khyber Pakhtunkhwa. We are grateful to the Alexander von Humboldt Foundation, Bonn, Germany, and the Higher Education Commission (HEC), Islamabad, Pakistan, for their financial support.
Funding
Naveed Wasif got the research support from the George Forster Fellowship (2015–2018) of the Alexander von Humboldt Foundation, Bonn, Germany. Saadullah Khan was supported through the NRPU project (No.4857/NRPU/R&D/HEC2014) by the Higher Education Commission (HEC), Islamabad, Pakistan. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SAK, MAK, NM, HB, NK, and NM have enrolled the patients and contributed to clinical diagnoses and report writing. NW has analyzed the exome sequencing data. NW and RY have contributed to Sanger Sequencing. SAK and SK have written the initial draft of the manuscript. NW and RY have critically reviewed and finalized the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Informed written consent was taken from affected and unaffected family members. All the participants were above 18-years of age. The work was approved by the Research and Ethical Committee of Kohat University of Science and Technology (KUST), Kohat, Pakistan.
Consent for publication
Along with consent for participation in the genetic study, informed written consent for publishing the clinical images and clinical information was obtained from all the participants.
Competing interests
The authors have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Khan, S.A., Khan, M.A., Muhammad, N. et al. A novel nonsense variant in SLC24A4 causing a rare form of amelogenesis imperfecta in a Pakistani family. BMC Med Genet 21, 97 (2020). https://doi.org/10.1186/s12881-020-01038-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01038-6