
Abstract
Objectives
Biallelic variants in solute carrier family 24 member 4 (SLC24A4) have been previously reported to cause non-syndromic autosomal recessive amelogenesis imperfecta (AI) of the pigmented hypomaturation type (MIM #615887). We here describe a novel variant in SLC24A4 causing mild enamel hypomaturation defects also in heterozygous individuals.
Materials and methods
In the present pedigree analysis, a large consanguineous Syrian family with AI of the hypomaturation type was investigated by clinical and dental evaluation, and exome and Sanger sequencing. Dental histological investigations of seven primary and two permanent teeth were performed.
Results
Homozygous variants in SLC24A4 (c.1604G>A; p.Gly535Asp) were identified in five individuals with brown discolorations and irregular pits and grooves of the teeth. Severe attritions, occlusal abfractions, and the radiological lack of contrast between enamel and dentin point out a mineralization defect. Histological dental investigations confirmed the clinical diagnosis of AI of the hypomaturation type. In two heterozygous individuals, a mild hypomaturation defect was present with white and light brown enamel discolorations.
Conclusions
This is the first report of heterozygous SLC24A4 variants causing mild hypomaturation defects, providing confirmatory evidence that the function of SLC24A4 in calcium transport has a crucial role in the maturation stage of amelogenesis.
Clinical relevance
The present report is expanding the clinical phenotype of SLC24A4 variants to more severe forms of amelogenesis imperfecta. An autosomal-dominant inheritance pattern with mild clinical phenotypes in heterozygotes has to be considered.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dental enamel is the most highly mineralized human tissue. It consists of a crystalline calcium phosphate (hydroxyapatite) and is characterized by the almost total absence of organic matrix [1]. Congenital disorders presenting with aberrations of enamel formation are denoted amelogenesis imperfecta (AI); prevalence ranges from 1:700 to 1:14000 within selected populations [2]. Enamel formation and growth are known to occur in three major developmental phases. Based on pathogenetic mechanisms and clinical features, AI is classified into three different types with at least 14 subtypes [3]. Hypoplastic AI is caused by disorders affecting the first step of amelogenesis, the synthesis of organic matrix. Impairment of enamel mineralization leads to hypocalcified AI, which is found only in a small number of AI patients. Disturbance of enamel maturation, the last step of amelogenesis, leads to hypomaturation AI. Clinical distinction of the major AI types and subtypes is difficult as subsequent developmental processes of enamel formation and growth are interdependent. For a clear diagnostic distinction, clinical and radiological specifications in connection with genetic information are of importance [4]. AI may be either non-syndromic, i.e., confined to the teeth, or syndromic, i.e., associated with various other clinical manifestations. It may be caused by pathogenic variants in various different genes with sometimes closely related functions. Inheritance may be autosomal dominant or recessive as well as X-linked [5] [6].
We here describe a large consanguineous family with a novel pathogenic missense variant in SLC24A4 causing non-syndromic AI. The gene product encodes a potassium-dependent sodium/calcium exchanger (NCKX4). In contrast to previous reports, heterozygous individuals showed a mild hypomaturation defect of the teeth, whereas AI in homozygotes was more severe than previously reported. We moreover present comprehensive clinical data together with extended histologic analyses of affected teeth paralleled by genetic analyses.
Material and methods
Ethical considerations
The study was conducted with the full understanding and written consent of all participants, or in cases of children, their parents as their legal guardians. The design and experimental analyses performed under the guidance of the biobank for rare diseases were approved by the Ethics Committee of the Medical University Innsbruck, Austria (Study No. UN4501) in accordance with the Helsinki Declaration of 1975 as revised in 2013.
Subjects and clinical investigations
A family displaced from Syria showing traits of AI in two generations (Fig. 1) was recruited in 2018 at the Department of Operative and Restorative Dentistry, Medical University of Innsbruck, Austria. Oral dental examination included visual and radiographic examination of teeth, as well as inspection and palpation of soft tissues. Blood samples were collected with full consent of the respective participant. All patients were screened by a general practitioner also providing further data by filling out a general medical questionnaire. If necessary, oral rehabilitation was provided at the department.

Pedigree of the family with missense variants c.1604G>A (p.Gly535Asp) in SLC24A4. Symbols: Males are marked as squares and females as circles. Black symbols mark individuals with homozygous missense variants and severe hypomaturated amelogenesis imperfecta. Gray symbols mark individuals with heterozygous missense variants and a mild phenotype. White symbols mark clinically non-affected individuals without a variant in SLC24A4
Histologic analysis
During oral rehabilitation of individuals I-2, II-1, II-4, II-5, and II-6, six teeth were extracted due to medical reasons and were available for histologic analysis. The teeth were dehydrated in solutions of increasing alcohol concentrations supplemented with basic fuchsin (Merck, Darmstadt, Germany) to accomplish block staining. The alcohol was then removed with acetone and the teeth were embedded in methyl methacrylate according to the manufacturer’s instructions (Heraeus, Technovit 9100 NEW). The embedded teeth were sawed perpendicular to the occlusal surface (Exact, Germany), in order to produce slices with a thickness of 25–30 μm which could then be polished (Exact, Germany) and contrast-stained with acetic light green (Light Green SF Yellowstich, Sigma Aldrich, Switzerland) [7].
Genetic studies
Genomic DNA was extracted from EDTA blood samples of both parents and their children using standard protocols. For massive-parallel sequencing, targeted genes exons and adjacent exon-flanking regions (± 15 bp) were enriched from isolated genomic DNA by Nextera® Rapid Capture (TruSightTM One Expanded Panel, Illumina, San Diego, CA) and sequenced on a NextSeq sequencing platform (Illumina, USA) using 2 × 150-bp paired-end chemistry in accordance with manufacturer’s instructions. Sequenced reads were aligned to the GRCh37/hg19 human reference. Data analysis including copy number variation (CNV) analysis was performed with the SeqNext®, and variants were evaluated with varSEAK® (JSI medical systems GmbH, Germany). Variant filtering was performed using the following criteria: (1) HPO term amelogenesis imperfecta (HP:0000705); (2) autosomal recessive inheritance; (3) MAF < 1%; (4) coverage > 20×; (5) stop, missense, and splice site variants; (6) not classified as benign or likely benign in variant database (ClinVar, in house). The classification of detected variants followed the consensus recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [8]. The SLC24A4 mutation c.1604G>A (NM_153646.3) was detected or confirmed in all family members by PCR amplification and Sanger sequencing from genomic DNA.
Results
Clinical features
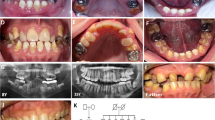
The propositus II:1 was a 12-year-old boy of a consanguineous family previously displaced from Syria to Austria presenting clinically with generalized hypomaturated AI (Fig. 2a). All teeth exhibited brown discoloration, irregular grooves, and pits on all teeth of primary and secondary dentitions. He presented an open bite. First molars, the only teeth with occlusal contacts, presented with occlusal abfractions. Dental panoramic radiography revealed the lack of contrast between enamel and dentin and agenesis of teeth 35 and 45 (Fig. 2b).

Intraoral photographs and radiograph of individuals with homozygous (a, b, e) or heterozygous (c, d) missense variants in SLC24A4 displaying hypomaturated AI. a Intraoral photograph and b panoramic X-ray of the propositus II-1 at 12 years of age, with homozygous missense variants. On the dental radiograph, enamel has the same radiodensity as dentin. Further radiologic findings are delayed eruption of tooth number 22, agenesis of teeth 35 and 45, and occlusal loss of substance of primary and secondary molars. Clinically, enamel of secondary teeth is hypomaturated with brownish discoloration and irregular grooves and pits. He presented a frontal open bite. c The teeth of the propositus’ mother I-1 at age 32 years and d individual II-2 at age 12 years with heterozygous missense variants. They present with whitish or light brown discolorations but smooth and hard enamel surfaces. Interdental gaps may be caused by mild enamel hypoplasia or occlusal discrepancies. e Clinical photograph of individual II-5 with homozygous missense variants. Intense yellow discoloration, rough enamel surfaces, and occlusal wear of primary and secondary teeth are present
His father I-2, aged 39 years, showed generalized brown discolored pits and grooves and cariogenic spots on the permanent teeth with softer enamel than normal. Premolars and molars were mostly provided with crowns and bridges. Missing 35 and 45 were suspiciously indicating agenesis of these teeth.
The propositus’ mother I-1, aged 32 years, showed a mild hypomaturated enamel with a white and light brown banding pattern (Fig. 2c). Interdental gaps in the upper jaw may be caused by mild enamel hypoplasia or occlusal discrepancies.
The sister of the propositus, individual II-2, presented at age 12 years with a complete permanent dentition and generalized interdental gaps. The enamel had normal hardness but a whitish smooth surface (Fig. 2d).
The second sister II-3, aged 10 years, presented with a mixed dentition and no signs of AI.
The brother, II-4, aged 8 years, showed a severe clinical phenotype of hypomaturated AI and a frontal open bite. Primary and secondary molars presented with severe occlusal chipping and attrition.
Sister II-5 at age 7 years exhibited a marked resemblance with individual II-4 bearing again a hypomaturated form of AI (Fig. 2e). Her teeth were very sensitive to temperature changes.
Clinical investigation of brother II-6 at age 5 years was incomplete due to lack of compliance. He also had an open bite, multiple cariogenic lesions, brownish discolorations of the teeth with pits and grooves. Dental radiograph displays a lack of contrast between enamel and dentin, and suspicious attrition or chipping of primary molars.
All pregnancies were reported uneventful and all deliveries were on term. During pregnancy, there was no abuse of medications, drugs, alcohol, or tobacco. All children were breastfed until the age of 12–24 months. Among other relatives, no known medical problems typically associated with AI such as nephrocalcinosis, platyspondylie, short stature, rod-cone retinal atrophy, no epilepsy or dementia, oncholysis, or bone anomalies were reported. All family members were free of abnormalities of hair, nails or skin, renal dysfunctions, or vision loss; were of normal stature; never experienced epilepsy; without hearing problems; and no abnormal bone fractures were reported.
Histologic analysis
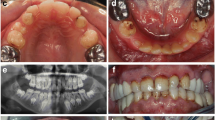
In total, seven primary teeth and two permanent teeth from individuals with homozygous missense variants in SLC24A4 were histologically analyzed. The sections showed that the enamel thickness was almost normal but the structure was immature. Defects with rough borders in the incisal/occlusal parts were seen in several teeth, reflecting chipping of the enamel (Fig. 3a). Retzius lines and Hunter-Schreger bands were missing in all specimens, and the enamel prisms were not regularly structured. Fractures and irregularities (prominent enamel tufts and an increased number of enamel lamels) were seen deep in the enamel (Fig. 3b, c). The thin surface zone had a normal mineralization, whereas the bulk of the enamel was hypomineralized in several specimens. The dentin had a normal structure. Based on clinical and histologic observations, AI in this family was classified as hypomaturation type.

Amelogenesis imperfecta in SLC24A4 is of the hypomaturation type. Histologic analysis of primary teeth extracted from individuals II-1, II-6, and a permanent tooth from I-2 is presented in this figure. The teeth were block stained with fuchsin, and after embedding in methylmethacrylate and perpendicular sectioning, the specimens were polished and contrast stained with acetic light green (original magnification 12–60×) [7]. a Overview of a ground section of a primary molar, showing an irregular enamel surface, abfraction, and structureless enamel. Chipping of enamel has occurred on the mesiobuccal cusp. The fuchsin-stained area (red) on the distal cusp is indicative for a caries lesion. The dentine has a normal structure. b Higher magnification of the mesiobuccal cusp. The abfraction is filled with debris. No Hunter-Schreger lines are visible; however, the neonatal line is distinguishable. There are a lot of cracks and enamel lamels and an increased number of tufts. c Transverse section of the enamel of a primary molar. The enamel shows a huge amount af cracks and lamellae. b Higher magnification of the mesiobuccal cusp. The abfraction is filled with debris. No Hunter-Schreger lines are visible; however, the neonatal line is distinguishable. There are a lot of cracks and enamel lamels and an increased number of tufts. c Transverse section of the enamel of a primary molar. The enamel shows a huge amount of cracks and lamellae
Genetic results
Exome sequencing in this consanguineous family revealed a homozygous missense variant c.1604G>A in SLC24A4 (NM_153646.3) located on 14q32.12 in all children presenting with severe hypomaturated AI and in their father (individuals I-2, II-1, II-4, II-5, II-6). Heterozygous carriers of the variant showed mild AI (mother I-1 and sibling II-2). The expected effect of this variant is an amino acid change p.Gly535Asp in the sodium/calcium exchanger membrane region of NCKX4 (Fig. 4). It concerns a highly conserved nucleotide (phyloP 6.19) and a highly conserved amino acid (up to fruit fly, considering 12 species). Prediction by AGVGD, SIFT, Mutation Taster, and PolyPhen-2 rate it as disease causing and damaging. The variant is not listed in gnomAD and thus is not a known polymorphism. The variant therefore is classified as probably pathogenic based on international criteria [8].

Predicted topology of full-length SLC24A4 (corresponding to transcript NM_153646.3, adapted from Parry et al. [10] including the known disease-causing variants (highlighted in orange) [9,10,11,12] and the here-detected variant (highlighted in red)). The variant c.1604G>A (p.Gly535Asp) lies in the sodium/calcium exchanger membrane region of SLC24A4 and is highly conserved in SLC24A4 orthologous. The conserved alpha repeats are shown in blue and green. The extracellular membrane is shown in gray
Discussion
Biallelic loss-of-function variants in SLC24A4 have been previously reported to cause non-syndromic autosomal recessive AI of the pigmented hypomaturation type, IIA5 (OMIM # 615887). The dental phenotype includes generalized yellow brown discoloration of smooth enamel surfaces, and a tendency for enamel chipping and attrition due to reduced enamel hardness [9,10,11,12]. Dental panoramic radiographs consistently show a lack of contrast between the enamel and dentin because of reduced mineral density [9,10,11,12].
Here, we report a novel SLC24A4 pathogenic missense variant (c.1604G>A; p.Gly535Asp) causing hypomaturated AI in two generations of a large consanguineous Syrian family. Homozygous individuals in this family had a more severe clinical phenotype than previously reported. They had an anterior open bite, which is usually a typical feature of hypomineralized AI, and generalized irregular enamel grooves and pits which could not be attributed to enamel chipping or fractures (Fig. 2b, c). In contrast to previous reports that describe heterozygous individuals as asymptomatic [9, 10], heterozygotes in the family reported here were symptomatic with whitish or light brown discolorations of the teeth, representing a mild enamel hypomaturation defect, but smooth and hard enamel surfaces (Fig. 2d, e).
SLC24A4 was first identified as a candidate gene involved in enamel maturation in 2012 by genome-wide transcript profiling of the developing enamel organ from rat incisors [13]. This gene encodes the solute carrier family 24 member 4 of the potassium-dependent sodium/calcium exchanger family. It is primarily expressed in ameloblasts during enamel maturation, when crystals expand in thickness, and the protein is thought to be a key Ca2+ transporter during amelogenesis [14]. SLC24A4 is not expressed in secretory-stage ameloblasts [12]. The important role of SLC24A4 in enamel maturation was further supported by animal and in vitro studies: SLC24A4−/− mice displayed enamel malformations and defects similar to those seen in humans [10], whereas transfection of HEK293 cells with different SLC24A4 variants resulted in abolition or at least a significant reduction in Ca2+ transport activity [15]. The proposed pathogenic mechanism is in accordance with the histologic findings in our study. The specimens showed an irregular structure of enamel prisms and an increased number of hypomineralized enamel tufts and lamels. In several sections, the bulk of the enamel was hypomineralized.
So far, eight individuals in five nuclear families have been reported with pathogenic mutations in SLC24A4 classified as null mutation, missense mutation, or exonal deletion (Fig. 4) [9,10,11,12]. Parry et al. reported on two individuals with AI from a Pakistani family carrying the SLC24A4 variant p.Ser499Cys [10], which is localized in the same protein domain as p.Gly535Asp. Additionally, Seymen et al. reported a consanguineous family carrying a 10-kb deletion in SLC24A4 gene [11] that impinges on sequence information for the sodium/calcium exchanger membrane, in which p.Gly535Asp is located. In contrast to these studies, we observed mild hypomaturation defects in heterozygotes. In most previous studies, clinical photographs of heterozygotes were not published, and mild hypomaturation defects may have been overlooked. Considering that the SLC24A4 protein functions as a monomer, it is unlikely that a dominant negative effect explains the development of mild clinical manifestation in the family studied by us. Most probably c.1604G>A (p.Gly535Asp) is a loss of function variant that either causes a substantial disturbance of the channel or impairs protein stability. Our observation indicates that half-normal transport capacities of the potassium-dependent sodium/calcium exchanger protein are not always sufficient for normal enamel mineralization at least in some individuals. Additional factors may influence local availability of calcium during amelogenesis, leading to clinically normal enamel if there is just enough calcium, or resulting in mild clinical phenotypes if there is just too little. Bronckers at el. suggested a role of the SLC24A4/NCKX4 Ca2+ transporter in the pathogenesis of fluorosis [16]. They proposed that the fluorotic effect is caused by reduced Ca2+ transport and as a consequence thereof lower mineral formation due to the decreased number of NCKX4 molecules incorporated into the apical membrane of ameloblasts. Indeed, the clinical phenotype of heterozygous individuals in our study appeared comparable with mild fluorosis.
The Enamel International Symposium called for more research on AI as there is a significant lag in the understanding of pathological mechanisms and phenotyping of affected tissues [17]. Deeper understanding regarding molecular targets and cellular mechanisms driving the development of healthy enamel may also provide new means of regenerative therapies and tissue engineering [17]. Besides challenging clinical issues, AI directly impacts the patient’s quality of life. Already mild phenotypes result in severe esthetic compromises. Cost coverage by health insurances and support groups is endorsed in cases with approved genetic etiology, while negligence as the cause of enamel defects was disproved.
The strength of the present pedigree analysis is the large number of included individuals and the accurate clinical, radiological, and genetic investigation of affected as well as mild or unaffected relatives. Detailed clinical data of heterozygotes are missing in most previous reports, making it nearly impossible to match them with our probands. There is one heterozygous sibling reported by Herzog et al. showing discrete white lines and spots on some teeth, which could be attributed to hypomaturation [9]. Reporting on only one nuclear family is a major limitation of the present study.
In conclusion, our study expands the list of pathogenic SLC24A4 variants and for the first time reports on mild hypomaturation defects in heterozygotes. Detailed histological evidence supports a crucial function of SLC24A4 for the provision of calcium in the maturation stage of amelogenesis. Future studies should focus on adapted acid-etching and bonding times of hypomaturated teeth to provide proper oral rehabilitation for affected individuals.
Change history
07 September 2020
In the pedigree, one of the individuals was marked as unaffected whereas it is heterozygous for the SLC24A4 mutation.
References
Nanci A (2008) Enamel: composition, formation, and structure. In: Nanci A (ed) Book title, 9th edn. Mosby Elsevier, St. Louis, USA
Crawford PJ, Aldred M, Bloch-Zupan A (2007) Amelogenesis imperfecta. Orphanet J Rare Dis 2:17. https://doi.org/10.1186/1750-1172-2-17
Witkop CJ Jr (1988) Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol 17:547–553
Poulsen S, Gjorup H, Haubek D, Haukali G, Hintze H, Lovschall H, Errboe M (2008) Amelogenesis imperfecta - a systematic literature review of associated dental and oro-facial abnormalities and their impact on patients. Acta Odontol Scand 66:193–199. https://doi.org/10.1080/00016350802192071
Wright JT, Carrion IA, Morris C (2015) The molecular basis of hereditary enamel defects in humans. J Dent Res 94:52–61. https://doi.org/10.1177/0022034514556708
Prasad MK, Laouina S, El Alloussi M, Dollfus H, Bloch-Zupan A (2016) Amelogenesis imperfecta: 1 family, 2 phenotypes, and 2 mutated genes. J Dent Res 95:1457–1463. https://doi.org/10.1177/0022034516663200
Stich H (1991) Oral Implantology. Thieme, Stuttgart
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Herzog CR, Reid BM, Seymen F, Koruyucu M, Tuna EB, Simmer JP, Hu JC (2015) Hypomaturation amelogenesis imperfecta caused by a novel SLC24A4 mutation. Oral Surg Oral Med Oral Pathol Oral Radiol 119:e77–e81. https://doi.org/10.1016/j.oooo.2014.09.003
Parry DA, Poulter JA, Logan CV, Brookes SJ, Jafri H, Ferguson CH, Anwari BM, Rashid Y, Zhao H, Johnson CA, Inglehearn CF, Mighell AJ (2013) Identification of mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger, as a cause of amelogenesis imperfecta. Am J Hum Genet 92:307–312. https://doi.org/10.1016/j.ajhg.2013.01.003
Seymen F, Lee KE, Tran Le CG, Yildirim M, Gencay K, Lee ZH and Kim JW (2014) Exonal deletion of SLC24A4 causes hypomaturation amelogenesis imperfecta. J Dent Res 93:366–370. doi: https://doi.org/10.1177/0022034514523786
Wang S, Choi M, Richardson AS, Reid BM, Seymen F, Yildirim M, Tuna E, Gencay K, Simmer JP, Hu JC (2014) STIM1 and SLC24A4 are critical for enamel maturation. J Dent Res 93:94S–100S. https://doi.org/10.1177/0022034514527971
Lacruz RS, Smith CE, Bringas P Jr, Chen YB, Smith SM, Snead ML, Kurtz I, Hacia JG, Hubbard MJ, Paine ML (2012) Identification of novel candidate genes involved in mineralization of dental enamel by genome-wide transcript profiling. J Cell Physiol 227:2264–2275. https://doi.org/10.1002/jcp.22965
Hu P, Lacruz RS, Smith CE, Smith SM, Kurtz I, Paine ML (2012) Expression of the sodium/calcium/potassium exchanger, NCKX4, in ameloblasts. Cells Tissues Organs 196:501–509. https://doi.org/10.1159/000337493
Jalloul AH, Rogasevskaia TP, Szerencsei RT, Schnetkamp PP (2016) A functional study of mutations in K+-dependent Na+-Ca2+ exchangers associated with amelogenesis imperfecta and non-syndromic Oculocutaneous Albinism. J Biol Chem 291:13113–13123. https://doi.org/10.1074/jbc.M116.728824
Bronckers AL, Lyaruu DM (2017) Magnesium, pH regulation and modulation by mouse ameloblasts exposed to fluoride. Bone 94:56–64. https://doi.org/10.1016/j.bone.2016.10.014
Kirkham J, Brookes SJ, Diekwisch TGH, Margolis HC, Berdal A, Hubbard MJ (2017) Enamel research: priorities and future directions. Front Physiol 8:513. https://doi.org/10.3389/fphys.2017.00513
Acknowledgments
We thank Günter Lepperdinger, Division of Genetics, University of Salzburg, Austria, for his support with microscopic analysis, and Isabel Hug, Shasta Pelzer, and Elvin Hasanov for the support with histologic sample preparations. We especially thank Gerda Ebner for organization of patients’ clinical oral investigation.
Funding
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study, or in cases of children, from their parents as their legal guardians.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 4003 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lepperdinger, U., Maurer, E., Witsch-Baumgartner, M. et al. Expanding the phenotype of hypomaturation amelogenesis imperfecta due to a novel SLC24A4 variant. Clin Oral Invest 24, 3519–3525 (2020). https://doi.org/10.1007/s00784-020-03222-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00784-020-03222-7