
Abstract
Background
Dyschromatosis symmetrica hereditaria (DSH) is a rare autosomal dominant cutaneous disorder caused by the mutations of adenosine deaminase acting on RNA1 (ADAR1) gene. We present a clinical and genetic study of seven unrelated families and two sporadic cases with DSH for mutations in the full coding sequence of ADAR1 gene.
Methods
ADAR1 gene was sequenced in seven unrelated families and two sporadic cases with DSH and 120 controls. Functional significance of the observed ADAR1 mutations was analyzed using PolyPhen 2, SIFT and DDIG-in.
Results
We describe six novel mutations of the ADAR1 gene in Chinese patients with DSH including a nonstop mutation p.Stop1227R, which was firstly reported in ADAR1 gene. In silico analysis proves that all the mutations reported here are pathogenic.
Conclusion
This study is useful for functional studies of the protein and to define a diagnostic strategy for mutation screening of the ADAR1 gene. A three-generation family exhibiting phenotypic variability with a single germline ADAR1 mutation suggests that chilblain might aggravate the clinical phenotypes of DSH.
Similar content being viewed by others


Background
Dyschromatosis symmetrica hereditaria (DSH; Mendelian Inheritance in Man no. 127400) is a rare pigmentary genodermatosis of autosomal dominant inheritance with high penetrance [1]. Ethnic background appears to be a major influence on the incidence of this disorder, as it is much more commonly found in Japanese and Chinese populations than in others [2]. RNA-specific adenosine deaminase 1 (ADAR1) gene, located on chromosome 1q21.3, was identified to be responsible for DSH [3]. In the present study, we performed a mutation analysis of the ADAR1 gene in seven Chinese families and two sporadic patients with typical DSH and identified six novel and two known mutations.
Methods
Ethics Statement
The current study conformed to the tenets of the Helsinki declaration and was approved by Ethical Committee of Shanghai skin disease hospital. All patients, their normal family members and 120 ethnically matched control individuals were informed about the purpose of the study and written consent was taken before recruitment and sampling. Informed written consent of minors was obtained from their guardians. Moreover, all the patients in our study gave their permission for publishing their images.
Clinical presentations
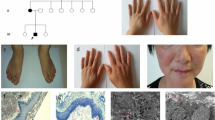
This study investigated 7 families (Family 1–7) and 2 sporadic cases with DSH in the Chinese population. DSH was diagnosed by experienced dermatologists based on the typical manifestations and histopathological findings. Seven multi-generation DSH families exhibited autosomal dominant inheritance. All affected individuals had a typical mixture of hyperpigmented and hypopigmented macules on the extremities (Fig. 1a, 1f). These lesions were of irregular shapes and sizes, which appears during early childhood, generally ranging from 3 to 15 years. Table 1 summarizes the clinical findings of all the patients analyzed in this study. Noteworthy is that a variable clinical phenotype was observed in family 7. The proband in family 7 was a 23-year-old female. She presented with asymptomatic hyperpigmented and hypopigmented macules on her extremities and had been developing freckle-like macules on her face since she was 7 years old (Figure 1b, g). Her mother has only a few small freckle-like pigmented macules disturbed on the back of her feet (Figure 1h, l). Whereas her uncle (II:3) had many more asymptomatic hyperpigmented and hypopigmented macules on his extremities than the proband but no freckle-like macules on his face (Figure 1c, i, m). There is no systematic involvement was noticed in any of our patients.

Clinical presentation of dyschromatosis symmetrica hereditaria (DSH) patients. a.. Freckle-like pigmented macules on the face (proband of family 3, severe phenotype). b. Freckle-like pigmented macules on the face of the proband in family 7. c. No freckle-like macules on the face of the patient II:3 in family 7. d. Dense freckle-like macules on the neck and face (proband of family 2, severe phenotype). e. Freckle-like pigmented macules on the face of the sporadic 2. f. Hypopigmented and hyperpigmented macules on the dorsal aspects of the extremities of hands and feet (proband of family 3, severe phenotype). g. Hypopigmented and hyperpigmented macules on the dorsal aspects of hands of the proband in family 7 (mild phenotype). h. No any lesion on the dorsal aspect of hand of the patient II:2 in family 7. i, j. Hypopigmented and hyperpigmented macules on the dorsal aspect of the hand (the patient II:3 in family 7 and the proband in family 2 , severe phenotype). k. Hypopigmented and hyperpigmented macules on the dorsal aspect of the hand of the sporadic 2. l. Few small freckle-like pigmented macules disturbed on the back of the foot (the patient II:2 in family 7). m, n. Hypopigmented and hyperpigmented macules on the back of the foot (the patient II:3 in family 7 and the proband in family 2, severe phenotype). o. Few small freckle-like pigmented macules disturbed on the back of the foot (the sporadic 2, mild phenotype)
Genetic studies
Polymerase chain reactions were performed as described previously [4]. In addition, samples from 120 unrelated population-match controls were sequenced for missense to exclude the possibility that these are polymorphism in the ADAR1 gene. Mutations were identified by comparing with the reported cDNA reference sequence (GenBank accession number: NM_001111).
Results
The entire coding and flanking intronic sequences of ADAR1 were screened for mutations among 7 families and 2 sporadic patients of Chinese origin with DSH, we detected six novel ADAR1 mutations and two previously described mutations by direct sequence analysis of the PCR products (Fig. 2). The spectrum of mutations included three nonsense mutations (p.R360X, p.R474X, p.W768X), one missense mutation p.R1083H, three frameshift mutations (p.Arg1030ThrfsX1036, p.Gly803ValfsX807, p.Asp1147ValfsX1184) and one nonstop mutation p.Stop1227R. We failed to detect any ADAR1 mutation in the sporadic 2.

Mutations of the ADAR1 gene found in patients with dyschromatosis symmetrica hereditaria. a. c.1078C>T (p.R360X) mutation in family 4. b. c.1420C>T (p.R474X) mutation in family 6. c. c.2303G>A (p.W768X) mutation in Sporadic 1. d.c.3248G>A (p.R1083H) mutation in family 7. e.c.3089_3090delGA (p.R1030DfsX1036) mutation in family 1. f. c.2408delG (p.G803VfsX807) mutation in family 3. g. c.3439ins17 (p.D1147VfsX1184) mutation in family 5. h. a nonstop mutation TGA(Stop) CGA(Arg)=Stop1227R in family 2
Three nonsense mutations were identified in the family 4, 6 and the sporadic 1. The mutation c.1078C > T (p.R360X) was detected in exon 2 in the family 4 (Fig. 2a). The resulted truncated protein will lack 867 amino acids. The other nonsense mutation c.1420C > T (p.R474X) was detected in exon 2 in the family 6 (Fig. 2b). The predicted protein lacked 753 amino acids. In the sporadic 1, a G > A mutation was found at nucleotide 2303 that generates a translational termination codon (Fig. 2c). The predicted protein lacked 459 amino acid residues.
One missense mutation c.3248G > A was found within exon 13 and was confirmed in the other patients and excluded in the remaining unaffected persons (Fig. 2d). This transition replaced a highly conserved arginine residue with histidine in codon 1083.
Three frameshift mutations were identified in the family 1, 3 and 5. Nucleotides GA were found to delete between c.3089 and c.3090 in the family 1 and this 2-bp small deletion, designated as p.Arg1030ThrfsX1036, induced a frameshift from codon 1030 and expected to produce a premature termination codon (PTC) at codon 1036 (Fig. 2e). One single-base deletion c.2408delG was identified within exon 7 in the family 3 and generated pre-terminating codon (PTC) at 4 codons downstream of deletion site (Fig. 2f). The mutation c.3439ins17 was found within exon 14 in family 5 and this 17-bp insertion generated a PTC at 37 codons downstream of insertion site (Fig. 2g). All the above frameshift mutations generated PTC respectively, and ADAR1 protein synthesis should end there without translating the full deaminase domain, which should produce inactive enzymes of ADAR1.
Moreover, a nonstop mutation in the normal stop codon 1227 of ADAR1 gene in exon 15 (TGA(Stop) ➙ CGA(Arg) = Stop1227R) (c.3679 T > C) was identified within exon 15 in family 2 (Fig. 2h). Theoretically, the Stop1227R mutation predictably results in an open reading frame and a mutant-type (MT) ADAR1 protein containing 1247 amino acid residues, compared with the 1226 amino acid residues of wild-type (WT) protein. The additional 21 amino acids (QYAPVTDGLGCVILGCERGRS) were included into the ADAR1 protein, with ensuing failure of formation of the healthy ADAR1 molecule and the development of DSH.
All the mutations except the nonstop mutation p.Stop1227R were predicted to be damaging. This prediction is based on the information from SIFT (http://sift.jcvi.org) [5], PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/) [6] and DDIG-in (http://sparks-lab.org/yueyang/server/ddig/) [7]. All the changes affect highly evolutionally conserved amino acids (Fig. 3).

All the mutations except the nonstop mutation p.Stop1227R were predicted to be damaging. a. Heatmap of PSSM for single mutations (p.R360X, p.R474X, p.W768X and p.R1083H). The red color indicates more conservation while blue indicates less conservation. b. Heatmap of PSSM for Indels (p.G803VfsX807, p.R1030DfsX1036 and p.D1147VfsX1184). The red color indicates more conservation while blue indicates less conservation.
Discussion
ADAR1, also called DSRAD (double-stranded RNA-specific adenosine deaminase), belongs to a family of RNA-specific adenosine deaminase that represents one type of RNA editing enzyme. It contains at least six functional domains: two Zalpha (Z-DNA-binding domain in adenosine deaminases), three DSRM (double-stranded RNA binding motif) and a tRNA-specific and double-stranded RNA adenosine deaminase (ADEAMc) domains. They are located in exon 2, exons 2–7 and exons 9–15, respectively [8].
In this study, we detected six novel ADAR1 mutations and two previously described mutations among 7 families and 2 sporadic patients of Chinese origin with DSH. The nonsense mutations (p.R360X, p.R474X and p.W768X) and single-base deletion c.2408delG (p.G803VfsX807) are expected to cause ADAR1 truncations lacking the entire ADEAMc domain while the other two frameshift mutations p.R1030TfsX1036 and p.D1147VfsX1184 in our study supposed to cause ADAR1 truncations lacking the partial ADEAMc domain, which should produce inactive enzymes of ADAR1 and ensue the development of this disease. All the frameshift indels were predicted to be deleterious by both SIFT and DDIG-in, all the nonsense mutations were predicted to be deleterious by DDIG-in and missense mutation c.3248G > A was predicted to be potentially damaging by both SIFT and Polyphen2.0 (Table 1). It is also possible that the mutant ADAR1 protein may not exist at all in these affected patients based on nonsense-mediated mRNA decay (NMD). NMD is a surveillance mechanism by which cells recognize and degrade mRNAs containing premature translation termination codons [9]. In family 5, we detected a nonsense mutation p.R474X, this mutation has been detected most frequently so far, and has been reported five times [3, 4, 10]. The hotspot mutation p.R474X has previously been reported in Chinese [4, 10] and also in Japanese [3] in many studies, indicating a possible founder effect of this mutation. No mutation was found in one sporadic 2 although he presents typical lesions of DSH (Fig. 1e, k, o). It is possible that the pathogenic mutation is regulatory ones within the promoter region or non coding regions. Such mutations will be missed in the current study where only the coding sequences and the exon-intron boundary sequences were targeted [4].
Of interest, we identified a nonstop mutation, c.3679 T > C (p.Stop1227R), in family 2. To our knowledge, this is a novel mutation and firstly reported in ADAR1 gene. Previous reports found that nonstop mRNA mutations associated with Diamond-Blackfan anemia [11], mucopolysaccharidosis II [12] and FX-coagulation deficiency [13] always result in pronounced reduction of steady-state levels of mutant mRNA. In contrast, quantitative real-time PCR and 3’-RACE-RFLP analysis revealed unreduced nonstop mRNA levels in the patient with mitochondrial neurogastrointestinal encephalomyopathy harboring a nonstop mRNA mutation (c.1416delC) in the TYMP gene [14]. However, we could not obtain experimental evidence because we lost contact with the patients in the family and could not get an additional blood sample for the quantitative real-time PCR and 3’-RACE-RFLP analysis. The proband in this family presented with severe phenotype including dense freckle-like macules on his neck and face, and multiple dense hyper- and hypopigmented lesions on the back of his hands and feet (Fig. 1d, j, n). Further functional studies are required to elucidate the pathomechanisms of the severe DSH underlying this nonstop mutation. Although still very much speculative, the following pathomechanisms of the severe DSH underlying this mutation may be considered: i) The p.Stop1227R mutation predictably results in an open reading frame and a mutant-type (MT) ADAR1 protein containing 1247 amino acid residues, compared with the 1226 amino acid residues of wild-type (WT) protein. The additional 21 amino acids (QYAPVTDGLGCVILGCERGRS) were included into the ADAR1 protein, with ensuing failure of formation of the healthy ADAR1 molecule. ii) It is likely that the mutant mRNA of ADAR1 gene in family 2 were reduced duing to nonstop mRNA decay, which was recently identified and showed to reduce the accumulation of abnormal transcripts lacking in-frame termination codons [11–13]. iii) The c.3679 T > C nonstop mRNA molecules are as stable as those transcribed from the wild-type alleles in this family but the translation of mutant transcript is repressed and/or its protein product is not stable [14].
Moreover, family 7 exhibits a phenotypic variability with a single germline mutation in ADAR1. The proband’s mother only has few small freckle-like pigmented macules disturbed on the back of her feet (Fig. 1l) whereas her uncle had much more asymptomatic hyperpigmented and hypopigmented macules on his extremities (Fig. 1i, m). According to his medical history, he used to have chilblain on his hands and feet during winter. Meanwhile, he had severe chilblain manifestated as blisters and erosions, which might have caused hypopigmented and hyperpigmented macules on his skin, which then aggravate the clinical presentation of DSH. In this family, we detected a missense mutation p.R1083H in ADAR1 gene, this mutation has been reported in the Japanese family and its patients are early-onset (under one year old) [15], while the proband in our family had her lesions since the age of 7. To sum up, we found that the same mutation could lead to different phenotypes even in the same family and did not establish a clear correlation between genotypes and phenotypes, thereby suggesting that phenotype is not explained by genotype alone and some factors, such as viral infection in utero and/or in infancy and exposure to ultraviolet light as well as chilblain in our patient, may affect the phenotype expression [1]. Li et al. compared the clinical features with the mutation identified in all families, but they also did not find a clear correlation between genotypes and phenotypes [4].
Conclusion
We have reported six novel mutations including a nonstop mutation p.Stop1227R, which was firstly reported in ADAR1 gene. Furthermore, we reported a three-generation family exhibiting phenotypic variability with a single germline ADAR1 mutation and chilblain might aggravate the clinical phenotypes of DSH. The ongoing recognition of different mutations may give insight into the still unknown mechanisms involved in the development of DSH.
Abbreviations
- ADAR1:
-
Adenosine deaminase acting on RNA1
- ADEAMc:
-
A tRNA-specific and double-stranded RNA adenosine deaminase
- DSH:
-
Dyschromatosis symmetrica hereditaria
- DSRAD:
-
Double-stranded RNA-specific adenosine deaminase
- DSRM:
-
Double-stranded RNA binding motif
- NMD:
-
Nonsense-mediated mRNA decay
- PTC:
-
Premature termination codon
- Zalpha:
-
Z-DNA-binding domain in adenosine deaminases
References
Hayashi M, Suzuki T. Dyschromatosis symmetrica hereditaria. J Dermatol. 2013;40(5):336–3.
Li M, Jin C, Yang L, Lai M, Yao Z. A novel complex insertion-deletion mutation in ADAR1 gene in a Chinese family with dyschromatosis symmetrica hereditaria. J Eur Acad Dermatol Venereol. 2011;25(6):743–6.
Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;73(3):693–9.
Li M, Yang L, Li C, Jin C, Lai M, Zhang G, et al. Mutational spectrum of the ADAR1 gene in dyschromatosis symmetrica hereditaria. Arch Dermatol Res. 2010;302(6):469–76.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Folkman L, Yang Y, Li Z, Stantic B, Sattar A, Mort M, et al. DDIG-in: detecting disease-causing genetic variations due to frameshifting indels and nonsense mutations employing sequence and structural properties at nucleotide and protein levels. Bioinformatics. 2015;31(10):1599–606.
Schade M, Turner CJ, Kühne R, Schmieder P, Lowenhaupt K, Herbert A, et al. The solution structure of the Zalpha domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc Natl Acad Sci U S A. 1999;96(22):12465–70.
Wagner E, Lykke-Andersen J. mRNA surveillance: the perfect persist. J Cell Sci. 2002;115(Pt 15):3033–8.
Zhang GL, Shi HJ, Shao MH, Li M, Mu HJ, Gu Y, et al. Mutations in the ADAR1 gene in Chinese families with dyschromatosis symmetrica hereditaria. Genet Mol Res. 2013;12(3):2794–9.
Chatr-Aryamontri A, Angelini M, Garelli E, Tchernia G, Ramenghi U, Dianzani I, et al. Nonsense-mediated and nonstop decay of ribosomal protein S19 mRNA in Diamond-Blackfan anemia. Hum Mutat. 2004;24:526–33.
Lualdi S, Di Rocco M, Corsolini F, Spada M, Bembi B, Cotugno G, et al. Identification of nine new IDS alleles in mucopolysaccharidosis II. Quantitative evaluation by real-time RT-PCR of mRNAs sensitive to nonsense-mediated and nonstop decay mechanisms. Biochim Biophys Acta. 2006;1762(6):478–84.
Ameri A, Machiah DK, Tran TT, Channell C, Crenshaw V, Fernstrom K, et al. A nonstop mutation in the factor (F) X gene of a severely haemorrhagic patient with complete absence of coagulation FX. Thromb Haemost. 2007;98(6):1165–9.
Torres-Torronteras J, Rodriguez-Palmero A, Pinós T, Accarino A, Andreu AL, Pintos-Morell G, et al. A novel nonstop mutation in TYMP does not induce nonstop mRNA decay in a MNGIE patient with severe neuropathy. Hum Mutat. 2011;32(4):E2061–8.
Kawaguchi M, Hayashi M, Murata I, Hozumi Y, Suzuki N, Ishii Y, et al. Eleven novel mutations of the ADAR1 gene in dyschromatosis symmetrica hereditaria. J Dermatol Sci. 2012;66(3):244–5.
Acknowledgements
We thank all members of the family for their cooperation in the study. This study was funded by grants from National Natural Science Foundation of China (81201222) and (81272990).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GLZ and MHS have carried out the sample collection, DNA isolation, PCR and sequencing, genotype and sequence analysis, sequencing alignment and drafted the manuscript. ZXL has carried out the statistical part and helped to draft the manuscript. GY and XFD have participated in the selection of studied patients and carried out diagnosis of patient. ML has participated in the study design, statistical analysis and drafted the manuscript. XLW has conceived the study, participated in its design, coordination and drafted the manuscript. All authors read and approved the final manuscript.
Ming Li and Xiu-li Wang, as first authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, G., Shao, M., Li, Z. et al. Genetic spectrum of dyschromatosis symmetrica hereditaria in Chinese patients including a novel nonstop mutation in ADAR1 gene. BMC Med Genet 17, 14 (2016). https://doi.org/10.1186/s12881-015-0255-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-015-0255-1