
Abstract
Dyskeratosis congenita (DC) is a telomeropathy presenting diagnostic and therapeutic challenges across multiple specialties; yet, subtle dermatological signs enable early detection, altering patient prognosis. A specific DC genetic sequencing was performed according to the clinical criteria of our patient in study. Subsequently, cross-checked information in the main genetic databases was carried out. Additionally, an extensive review of the literature was made to organize the main dermatological aspects in DC. We report a novel variant of DC. Additionally, we share 10 useful and practical messages for dermatologists and any specialist caring for this group of patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dyskeratosis congenita and disorders related to telomere biology (DC/TBD) are caused by poor telomere maintenance resulting in short telomeres [1, 2]. The prevalence of this telomeropathy is 1/1,000,000,000 with a male predominance (13:1) [3, 4]. Given the limited information that exists it can be misdiagnosed. Approximately 9 cases per year have been described from 1910 to 2022 in which the genes ACD, CTC1, DKC1, NHP2, NOP10, PARN, RTEL1, TERC, TERT, TINF2 and WRAP53 are implicated with DC/TBD [5].
This study highlights dermatologic diagnostic clues useful for healthcare professionals to suspect it and how these findings influence genetic testing requests. Additionally, a novel mutation in the TERT gene is reported.
Methods and case
11-year-old boy with a 9-year history of progressive mucosal and adnexal changes. At 3 years old, he developed tongue architecture alterations, including a tumor-like plaque with a velvety, whitish surface on the left lateral and posterior side, variable nail changes, and light brown reticular hyperkeratosis on the palms. Additional findings included short stature, developmental delay, and periodontal disease.
At 7 years old, he presented with esophageal stenosis, rectal fistula, bone marrow aplasia, and recurrent thrombocytopenia, requiring multiple corticosteroid and cyclosporine treatments. Family history revealed a sister who died of bone marrow aplasia at 17, while parents had no phenotypic abnormalities. Differential diagnoses included 20-nail dystrophy, Nail-patella syndrome, DC, poikiloderma with neutropenia, and other ectodermal dysplasias. Hematological findings raised suspicions of Fanconi anemia, Diamond-Blackfan anemia, and Shwachman-Diamond syndrome.
We considered it unnecessary to perform an exome by mass sequencing, and therefore targeted genetic sequencing was performed in DC/TBD [5]. From genomic DNA extraction, the exome library was prepared by probe capture and PCR enrichment of coding regions and intronic regions (+ 20 bp) adjacent to the exons of approximately 21 thousand genes (Agilent SureSelect Human All Exome V6). The exome library was fed into the Illumina NovaSeq-6000 sequencer and the results were analyzed based on the human genome reference sequence (hg19) and subsequently (hg38) by Varsome [6]. Sequencing, annotation, and variant calling, were performed in Novogene Sample Receiving Admera Health and bioinformatics analysis in collaboration with Sophia Genetics DDM, meeting specific quality criteria. Coverage > 90% and an average depth of 103x were obtained. The targeted panel/exome results only contain findings related to the patient’s clinical history and are analyzed considering OMIM, Ensembl, LOVD, HGMD, ClinVar, gnomAD, Varsome, Beacon Network databases, in silico prediction tools, and American College of Medical Genetics (ACMG) variant classification criteria.
Results
We identified two variants in the TERT gene (Fig. 1) with uncertain clinical significance (VUS) both in the heterozygous state. The c.2707 A > G variant is a new variant since it has not been reported in the Ensembl, RefSeq, gnomAD, ClinVar, Varsome, or Beacon Network databases. This variant has a strong clinical-genetic correlation with predictors of pathogenicity based on evidence of multiple predictors in silico. The Meta Score by varsome was 10 for moderate pathogenicity (BayesDeladdAF, MetaLR, MetaRNN, MetaSVM, REVEL) and 9 individual predictors for moderate pathogenic variant (CADD, Polyphen2, HDIV, Polyphen2 HAVR, DEOGEN2, EVE, M-CAP) with 3 supporting predictors of pathogenicity (FATHMM, mutation assessor, MVP) (see attached files) [6]. Regarding the c.1663G > A variant, it has already been described in the literature and has not been the subject of our study.

Molecular characteristics of the novel variant. Panel A shows the telomerase-shelterin complex which is composed of the H/ACA-snoRN complex (dyskerin, NOP10, NHP2 and GAR1,), TERC consisting of a functional non-coding RNA molecule, composed of 3 domains: 1. Pseudonucleotide, responsible for transporting the nucleotide template. 2. CR4-CR5 region, where it binds to TERT. 3. 3’ terminal, responsible for binding to the proteins of the H/ACA-snoRN complex and finally the Shelterin complex composed of its 6 protein subunits TRF1, TRF2, TPP1, POT1, RAP1 and TIN2, the latter responsible for the stabilization of the telomerase-shleterin complex [21]. Panel B shows the molecular analysis of the patient in study. There are two missense variants with uncertain clinical significance c.2707 A > G (TERT: NM_198253, p.Thr903Ala), and c.1663G > A (TERT: NM_198253,p.Glu555Lys) both in the heterozygous state. Panel C shows a schematic diagram of the variant c.2707 A > G taken from Varsome® which describes different characteristics of this novel variant mutation: ACGT, Transcripts, Pathogenicity and Variants. Our variant in study is the one located in the thymine (T) base chr5: 1,264,540 in the Threonine amino acid. The variant which is located inside the blue box is classified as LP. Note that the variant has not been described before in databases in comparison with other previous variants [6]. P: Pathogenic LP: Likely Pathogenic VUS: Uncertain Clinical Significance LB: Likely Benign B: Benign.
Discussion
Clinical workup and update
This genetic disease is defined by the classic triad: dystrophic nails, oral leukoplakia, and reticular pigmentation in the upper thorax and/or neck [7]. Only a portion of DC patients exhibit the full triad, with approximately 80–90% presenting at least one mucocutaneous manifestation [2]. Some patients may also develop systemic complications as subtle phenotypic characteristics emerge gradually [7].
Ungular findings
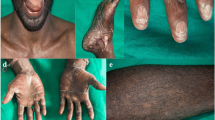
The involvement can be ungular or periungual (atrophy of the fingertips). Nail findings include anonychia, onychatrophia, onychoschizia, ungueal striae, longitudinal ridges, trachyonychia, koilonychia, ventral and dorsal pterygium, boxnail pattern, clubbing nails, leukonychia and black chromonychia [4, 7]. These findings are reported in up to 88% of cases [3]. Additionally, we found subungual and periungual hemorrhages that give clues of hematologic involvement (Fig. 2).
Mucosal findings
Mucocutaneous features typically develop between 5 and 15 years of age [3]. The main characteristic is leukoplakia involving the tongue (Fig. 2), oral mucosa, gums, or hard palate (60–80%), with a potential for malignant transformation and it can be detected in the first months of life [3, 8]. Less frequent manifestations are lichen planus, brown pigmentation, papillary and mucosa atrophy, short blunted roots, hypocalcification, taurodontism, periodontitis, early caries, gingival edema and hemorrhage [8]. Histopathology reveals hyperplasia and parakeratotic hyperkeratosis with variable dysplastic changes [4]. The involvement of other mucous membranes includes dysphonia, dysuria, phimosis, epiphora, stenosis of the lacrimal drainage system, blepharitis, ectropion, and entropion [3].
Skin and other adnexal manifestations
In most cases, patients present with grayish-brown hyperpigmentation, or in a minority of cases hypopigmentation, which may be subtle or diffuse, associated with atrophy, and poikiloderma [2,3,4]. The localization of the lesions usually appears in sun-exposed or flexural areas [4].
Other manifestations include palmoplantar hyperkeratosis (Fig. 2), blistering with trauma, chronic ulceration of the fingers and hyperhidrosis prevalent in 15% of cases, which may involve the palmoplantar regions, forehead, or axillae [4, 9]. Hair is also affected leading to fine appearance, alopecia of the scalp, loss of eyebrows or eyelashes, premature graying and trichiasis [4, 10].
Risk of malignant transformation
Telomere shortening increases chromosomal abnormalities, elevating the risk of malignant transformation, with oral mucosa squamous cell carcinoma (SCC) being the most common in DC [10]. SCC develops from oral leukoplakia or erythroleukoplakia of long evolution. Malignant transformation is between 0.13% and 34%, according to the clinical presentation which may be thin, thick/homogeneous, granular, verruciform, or erythroleukoplakic [5, 11, 12]. Less frequently, there is a risk of developing SCC in the nasopharyngeal mucosa, esophagus, vagina, cervix, and rectum [5, 10, 13]. Skin SCCs most commonly occur on the head and neck but can appear anywhere, particularly in sun exposed areas or in patients receiving post-Bone Medular Transplantation (BMT) therapy [10].
People with DC tend to develop these malignant tumors at an earlier age compared to the general population [1, 10]. The average age of diagnosis of SCC is between 20 and 30 years, where most of the reports are concentrated before a BMT, for basal cell carcinoma, its report is restricted to events after BMT, being less frequent [5].
Graft-versus-host disease
BMT recipients are at risk of developing cutaneous graft versus host disease (GVHD), mimicking poikiloderma and nail changes seen in dyskeratosis. Chronic GVHD initially presents as violaceous erythema, progressing to lichen planus or sclerodermiform lesions. This inflammatory condition, often alongside hyperpigmentation, blistering, nail dystrophy, and alopecia, complicates the differential diagnosis with DC [10, 14]. The suspicion of acute or chronic GVHD should be presumed according to generalized skin involvement after BMT and histopathological support [10, 14, 15]. Our patient has not yet received BMT.
Biomolecular background
The diagnostic suspicion of DC is clinical, and its confirmation is molecular [7]. Vulliamy et al. proposed diagnostic criteria for DC, categorized into major (classic triad) and minor criteria, detailed in Table 17. Our patient meets two criteria of the classic triad and five of the minor criteria.
The presentations of this telomeropathy arise through recessive inheritance, heterogeneous dominant, X-linked, or by de novo mutation generating direct effects on telomerase [16]. In our case, we found an autosomal recessive inheritance. The proportion of telomeropathies associated with DC due to TERT is described between 1 and 7% for autosomal dominant and/or recessive forms [17,18,19,20]. Telomerase is a holoenzyme that is responsible for maintaining telomere length of stem and germ cell chromosomes, preventing cellular senescence and apoptosis [21].
Telomerase consists of a ribonucleic component (TERC) with 451 ribonucleotides, acting as a template for adding TTAGGG to telomeres’ 3’ ends for elongation, and an enzymatic component, telomerase reverse transcriptase (TERT) (Fig. 1) [21]. This complex promotes the synthesis and stabilization of TERC, favoring telomeric genesis [21, 22].
Mutations in the TERC gene are believed to be an important cause of the occurrence of the autosomal dominant form of dyskeratosis congenita [7, 15, 16, 22,23,24]. DKC1 (dyskerin) is the most frequently reported mutation (20-25%) in DC, in contrast to the 1–7% described in the TERT gene, highlighting this rare case [7].
The variants identified in this case, c.2707 A > G and c.1663G > A, both in heterozygous state in the TERT gene (5p15.33) are associated with autosomal dominant (AD) and autosomal recessive (AR) dyskeratosis congenita, pulmonary fibrosis and/or bone marrow failure, telomere-related (AD) and acute myeloid leukemia (somatic and AD) (OMIM: 187,270). The c.2707 A > G variant is not reported in the public databases Ensembl, RefSeq, gnomAD, ClinVar, or Varsome nor the literature, its minor allele frequency (MAF) is estimated to be less than 1%, suggesting that the described change may have a negative effect on the phenotype due to its low frequency in the general population [6]. This is a missense variant whose DNA change results in the substitution of the amino acid threonine for alanine, which are residues with different physicochemical properties [6]. The p.Thr903Ala change occurs at a moderately conserved position among species (PhyloP) and 17 of 19 bioinformatic predictors classify it as pathogenic (Varsome) [6]. Therefore, the structure/function of the protein could be affected. Varsome PP2 helps support this variant as probably pathogenic [6]. These scores and rules follow the ACMG and the bioinformatic predictors estimated that it could have a pathogenic effect with a high probability and point to a de Novo mutation (see attached files) [6, 25].
Regarding both variants being missense mutations, based on the ACMG guidelines, since benign missense mutations are rare, a new missense mutation can be seen as evidence supporting its potential to cause disease, with a likelihood of being moderately or highly pathogenic [25].
Both identified variants are VUS and require classification. Determining their phase (compound heterozygosity or simple heterozygosity) is crucial, suggesting future parental molecular analysis to elucidate their role in DC development. Nevertheless, genetic predictor results and clinical features strongly indicate pathogenicity in this case.
Limitations and precisions
Our methodology’s limitations include focusing on point mutations in exonic or splicing regions, and small deletions or insertions. Large chromosomal rearrangements, deep intronic mutations, and variants in regulatory regions may be missed. Additionally, variants in unanalyzed or repeat expansion regions, as well as low-frequency mosaicisms, may not be reliably detected. Only pathogenic, possibly pathogenic, or VUS variants are reported; benign variants are excluded. Variant classification may change with scientific advances. A negative result doesn’t rule out untested mutations.
Conclusions and highlights
This novel TERT gene variant and its dermatologic manifestations represent the first unreported case globally, making it a valuable addition to databases. We provide 10 practical messages for dermatologists and specialists caring for these patients.
(I) All patients aged 5–15 with confirmed bone marrow aplasia should undergo dermatological assessment for potential genodermatosis manifestations. (II) Given the risk of periodontal disease, caries, and oral malignant neoplasms, close follow-up every 6 months by dentistry is recommended. (III) If the patient undergoes BMT, sources of latent oral infection of dental and periodontal origin should be eliminated. (IV) An annual evaluation to rule out neoplasms, stenosis, hepatic, pulmonary fibrosis, and corneal abrasions is recommended. (V) Simple blood tests are suggested every 6 months to look for bone marrow aplasia. (VI) Early hematologic involvement should be suspected in subjects with subungual and periungual hemorrhages. (VII) In all patients older than 20 years, SCC should be ruled out. (VIII) Dermatologists should be trained in the recognition of acute and chronic manifestations associated with GVHD. (IX) Patients must avoid carcinogenic activities like smoking, alcohol consumption, and unprotected sun exposure; prioritizing photoprotection with physical and chemical measures, even indoors. (X) Not all patients comply with the typical localization of the classic triad so a thorough physical examination by dermatology should be performed [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25].

Clinical findings in DC (a) Onychodystrophy; (b) Trachyonychia; (c) Koilonychia; (d) Ventral pterygium (e) “Boxnail” pattern; (f) palmar hyperkeratosis; (g) Oral hairy leukoplakia on the tongue. Note subungual and periungual hemorrhages in (c) and (d) that give clues of hematologic involvement
Data availability
Data is provided within the related files.
References
Brailovski E, Tsui H, Chen Y, Bin, Velsher L, Liu J, Buckstein R Previously unreported WRAP53 gene variants in a patient with dyskeratosis congenita. Ann Hematol [Internet]. 2022 Apr 1 [cited 2023 Feb 14];101(4):907–9. https://link.springer.com/article/https://doi.org/10.1007/s00277-021-04678-7
Barbaro PM, Ziegler DS, Reddel RR (2016) The wide-ranging clinical implications of the short telomere syndromes. Intern Med J 46(4):393–403
Kumar S, Suthar R, Dyskeratosis, Congenita (2013) JK Sci 15(2):56–58
AlSabbagh MM (2020) Dyskeratosis congenita: a literature review. JDDG - J German Soc Dermatology 18(9):943–967
Alter BP, Giri N, Savage SA, Rosenberg PS (2009) Cancer in dyskeratosis congenita. Blood 113(26):6549–6557
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R et al VarSome: the human genomic variant search engine. Bioinformatics [Internet]. 2019 Jun 1 [cited 2023 Dec 3];35(11):1978–80. https://doi.org/10.1093/bioinformatics/bty897
Savage SA (2020) Dyskeratosis Congenita Summary Genetic counseling GeneReview Scope. ;1–30
Koruyucu M, Barlak P, Seymen F (2014) Oral and dental findings of dyskeratosis congenita. Case Rep Dent [Internet]. [cited 2023 Dec 3];2014:1–5. https://pubmed.ncbi.nlm.nih.gov/25610666/
Reimann C, Kloeckener-Gruissem B, Niemeyer CM, Vanscheidt W Late manifestation of dyskeratosis congenita presenting as chronic dermal ulcer in a 37-year-old man. Journal of the European Academy of Dermatology and Venereology [Internet]. 2008 Jul 1 [cited 2023 Dec 3];22(7):897–8. https://onlinelibrary.wiley.com/doi/full/https://doi.org/10.1111/j.1468-3083.2007.02530.x
Savage SA, Cook EF (2015) Dyskeratosis Congenita and Telomere Biology Disorders: Diagnosis and Management Guidelines. Dyskeratosis Congenita Outreach, Inc [Internet]. ; https://www.dcoutreach.org/guidelines
Warnakulasuriya S, Ariyawardana A Malignant transformation of oral leukoplakia: a systematic review of observational studies. Journal of Oral Pathology & Medicine [Internet]. 2016 Mar 1 [cited 2023 Dec 3];45(3):155–66. https://onlinelibrary.wiley.com/doi/full/https://doi.org/10.1111/jop.12339
Masthan KMK, Babu NA, Sankari SL, Priyadharsini C, Leukoplakia A short review on malignant potential. J Pharm Bioallied Sci [Internet]. 2015 Apr 1 [cited 2023 Dec 3];7(Suppl 1):S165. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4439659/
Cherian LM, Deepthi K, Rejani ER (2010) Dyskeratosis Congenita; a Case Report and Review of Literature. Oral Maxillofacial Pathol J. ;1(2)
Treister N, Lehmann LE, Cherrick I, Guinan EC, Woo S, Bin Dyskeratosis congenita vs. chronic graft versus host disease: Report of a case and a review of the literature. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontology [Internet]. 2004 Nov 1 [cited 2023 Dec 3];98(5):566–71. http://www.oooojournal.net/article/S1079210404000678/fulltext
Soledad Fernández García M, Teruya-Feldstein J (2014) Journal of Blood Medicine Dovepress The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med [Internet]. [cited 2023 Feb 24];5–157. https://doi.org/10.2147/JBM.S47437
Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I (2006) Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 107(7):2680–2685
Ballew BJ, Savage SA Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol [Internet]. 2013 Jun [cited 2023 Dec 3];6(3):327–37. https://www.tandfonline.com/doi/abs/https://doi.org/10.1586/ehm.13.23
Dokal I, Vulliamy T, Mason P, Bessler M Clinical utility gene card for: Dyskeratosis congenita – update 2015. European Journal of Human Genetics 2015 23:4 [Internet]. 2014 Sep 3 [cited 2023 Dec 3];23(4):558–558. https://www.nature.com/articles/ejhg2014170
Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA Unraveling the pathogenesis of Hoyeraal–Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol [Internet]. 2015 Aug 1 [cited 2023 Dec 3];170(4):457–71. https://onlinelibrary.wiley.com/doi/full/https://doi.org/10.1111/bjh.13442
Bertuch AA (2016) The molecular genetics of the telomere biology disorders. RNA Biol [Internet]. Aug 2 [cited 2023 Dec 3];13(8):696–706. https://www.tandfonline.com/doi/abs/https://doi.org/10.1080/15476286.2015.1094596
Kirwan M, Dokal I (2009) Dyskeratosis congenita, stem cells and telomeres. Biochimica et Biophysica Acta (BBA) -. Mol Basis Disease 1792(4):371–379
Grill S, Nandakumar J (2021) Molecular mechanisms of telomere biology disorders. J Biol Chem [Internet]. Jan 1 [cited 2023 Nov 7];296:100064. Available from: /pmc/articles/PMC7948428/
Perona R, Sastre L, Callea M, Clinical (2020) Etiological Therapeutic Aspects Dyskeratosis Congenita 4(1):77–82
Ratnasamy V, Navaneethakrishnan S, Sirisena ND, Grüning NM, Brandau O, Thirunavukarasu K et al (2018) Dyskeratosis congenita with a novel genetic variant in the DKC1 gene: a case report. BMC Med Genet 19(1):4–9
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med [Internet]. May 8 [cited 2024 Jan 27];17(5):405–24. https://pubmed.ncbi.nlm.nih.gov/25741868/
Funding
Not applicable.
Open Access funding provided by Colombia Consortium
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by C.N.M, D.C, I.F and L.C.R. The first draft of the manuscript was written by C.N.M, D.C, F.A, F.I.U, P.A.R and all authors commented on previous versions of the manuscript. Figures 1 and 2 were prepared by C.N.M, D.C and F.A. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Approved by Universidad de La Sabana’s Institutional Ethics Committee. Techniques followed the 1975 Declaration of Helsinki. Informed consent obtained from patient’s parents, approving photo anonymity.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neri Morales, C., Cuestas, D., Ángel, F. et al. Dyskeratosis congenita associated with a novel missense variant in TERT: Approach for the dermatologists. Arch Dermatol Res 316, 438 (2024). https://doi.org/10.1007/s00403-024-03050-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00403-024-03050-9