
Abstract
Background
Imerslund-Gräsbeck Syndrome (IGS) is a rare autosomal recessive disease characterized by intestinal vitamin B12 malabsorption. Clinical features include megaloblastic anemia, recurrent infections, failure to thrive, and proteinuria. Recessive mutations in cubilin (CUBN) and in amnionless (AMN) have been shown to cause IGS. To date, there are only about 300 cases described worldwide with only 37 different mutations found in CUBN and 30 different in the AMN gene.
Case presentation
We collected pedigree structure, clinical data, and DNA samples from 2 Caucasian English half-sisters with IGS. Molecular diagnostics was performed by direct Sanger sequencing of all 62 exons of the CUBN gene and 12 exons of the AMN gene. Because of lack of parental DNA, cloning, and sequencing of multiple plasmid clones was performed to assess the allele of identified mutations. Genetic characterization revealed 2 novel compound heterozygous AMN mutations in both half-sisters with IGS. Trans-configuration of the mutations was confirmed.
Conclusion
We have identified novel compound heterozygous mutations in AMN in a family from the United Kingdom with clinical features of Imerslund-Gräsbeck Syndrome.
Similar content being viewed by others


Background
Imerslund-Gräsbeck Syndrome (IGS) is a rare autosomal recessive disorder characterized by intestinal cobalamin (vitamin B12) malabsorption usually leading to megaloblastic anemia in childhood. IGS was first described in 1960 by two physicians, Olga Imerslund from Norway [1] and Ralph Gräsbeck from Finland [2]. Clinical features of IGS include failure to thrive, recurrent infections, anemia, and mild proteinuria. IGS should also be considered in individuals with intermittent nephrotic-range proteinuria [3].
Genetic investigations in affected IGS families from Finland identified bi-allelic mutations in the gene CUBN, encoding cubilin [4] whilst in Norwegian families mutations in AMN, encoding amnionless were found [5]. Since then, many additional genetically confirmed cases have been reported with about 300 IGS cases published worldwide [6]. IGS is thought to have a prevalence of about 1/200,000 with a geographical concentration in Scandinavia and the Middle East [7]. Most of the published cases have been identified in Finland, Norway and eastern Mediterranean countries frequently in consanguineous families [8]. To date, only 37 different mutations in CUBN and 30 different mutations in AMN have been reported in the “Human Gene Mutation Database” (HGMD) worldwide (http://www.hgmd.cf.ac.uk/ac/index.php).
Cubilin and amnionless are required for intestinal absorption of intrinsic factor-vitamin B12 complex and renal proximal tubular absorption of filtered plasma proteins. Cubilin is a large (460 kDa) membrane-associated multiligand receptor and amnionless is a 50 kDa type 1 transmembrane protein [9]. Together they function as a heterodimeric complex known as cubam [10]. Both proteins are highly expressed in the small intestine and proximal tubule of the kidney. Within the proximal tubule they interact with megalin (a multi-ligand binding receptor encoded by LRP2), allowing reabsorption of low molecular weight (LMW) proteins including albumin [11]. Mutations in GIF, encoding gastric intrinsic factor may give a similar hematological and clinical picture, but this is classed as a hereditary intrinsic factor deficiency and should not be confused with IGS [12].
The uptake of LMW proteins within in the kidney initially involves filtration in the glomerulus and then reabsorption within the tubules [13]. Almost all the filtered plasma proteins are reabsorbed in the renal proximal tubule by receptor-mediated endocytosis. The only identified receptors mediating this endocytosis are megalin and cubilin. There are multiple recognised ligands for cubilin and megalin [14]. Once the ligand binds to the receptor at the apical plasma membrane, it is internalised via a vesicle. Acidification within the intra-vesicular lumen then causes the receptor and ligand to separate. The ligand is then transported to its required location and the receptor recycled back into the luminal membrane. Studies in murine models indicate that within the tubule, megalin functions mainly to facilitate the internalisation of the cubilin–albumin complex [15]. As the normal functioning of cubulin is dependent on amnionless, defects in either cubilin or amnionless result in reduced protein reabsorption and subsequent LMW proteinuria. In IGS, published cases have suggested that the proteinuria is non-progressive and has not been associated with clinical kidney disease hence renal function is preserved [16]. Although vitamin B12 replacement corrects haematological parameters, proteinuria usually persists.
Interestingly there have been cases of confirmed IGS, where LMW proteinuria is not present. Currently no clear link has been identified to associate specific mutations to clinical correlations, however with increasing identification of mutations, it appears that functional null mutations in the cubilin or amnionless gene are more likely to be associated with proteinuria than those with a missense mutation [17].
Here we report novel compound heterozygous mutations in AMN in 2 half-sisters from the United Kingdom.
Case presentation
We identified a Caucasian English family in whom two half-sisters had a clinical diagnosis of IGS (Fig. 1). In both cases, clinical presentation had been at an early age. The eldest sister (III:1) was diagnosed with pernicious anemia at the age of 2 years and was commenced and continued on vitamin B12 supplementation since then. Sub-nephrotic range proteinuria was first identified on urine dipstix testing during her teenage years but was not formally quantified until her first pregnancy at 33 years of age. Her proteinuria was non progressive, and was not associated with hypertension or pre-eclampsia and persisted at similar levels (0.6–0.7 g/24 h) post-natally. Similarly, the younger sister (III:2) also presented with anemia aged 3 years and has been successfully managed with vitamin B12 supplementation. In patient III:2, proteinuria was also first identified at 10 years of age and then quantified (at a sub-nephrotic range) during her pregnancy (age 23 years) and has persisted at similar levels since then. Proteinuria was documented at 0.5 g/24 h at 36 weeks gestation and by delivery (38 weeks gestation) proteinuria had reached 0.9 g/24 h, without the development of systolic hypertension. Postpartum she continued to have persisting sub-nephrotic range proteinuria (0.7 g/24 h). A summary of clinical details is provided in Table 1.

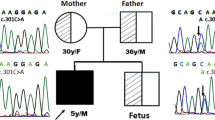
Pedigree diagram of family and chromatograms of AMN mutations in a family with Imerslund-Gräsbeck syndrome. a Pedigree diagram of a sibship with Imerslund-Gräsbeck syndrome. Circles represent females, squares represent males. Full symbols denote patients with IGS, dots equals presumed carrier status. b Chromatograms of 2 novel compound heterozygous mutations in the gene AMN (Genbank NM_030943.3), encoding for the protein amnionless, identified in 2 half-sisters (III-1 and III-2) with Imerslund-Gräsbeck syndrome
Both sisters were commenced on 3 monthly vitamin B12 supplementation in early childhood and abnormalities of growth or developmental were not documented. There have been no identified neurological abnormalities in either sibling however multiple urinary tract infections were reported and documented in both sisters throughout their teenage years and into adulthood. The half-sisters continue to receive B12 supplementation however, despite well maintained haemoglobin levels, both siblings continue to experience symptoms of lethargy and tiredness. Sister III:1 became anemic during her pregnancy and required extra B12 supplementation along with iron supplementation.
Careful review of the family history revealed that each of the half-sisters’ fathers were brothers, which was compatible with an autosomal recessive pattern of disease (Fig. 1a). We therefore examined the underlying genetic cause of IGS in this family by performing direct Sanger sequencing of the genes AMN (12 exons) and CUBN (67 exons) known to be implicated in autosomal recessive IGS. Mutation analysis revealed two novel compound heterozygous mutations in the AMN gene in both affected half-sisters. We identified a heterozygous deletion of an adenine nucleotide in exon 1, leading to a frame shift and predicted premature stop 5 codons downstream (c.35delA, p.Gln12Argfs*5) and we found a heterozygous missense mutation (c.206 T > A, p.Met69Lys) in exon 3 of the AMN gene (Fig. 1b). Both mutations have not been reported in any public database including “The Exome Aggregation Consortium” (ExAC) data set with exome sequence data obtained from 63,352 individuals (http://exac.broadinstitute.org/).
Since the patients’ mother was deceased and both fathers were uncontactable, no parental DNA material was available for segregation analysis of the mutations. To determine whether the mutations were inherited in cis or trans, we amplified and cloned the respective genomic region encompassing the location of both identified AMN mutations. We successfully amplified and cloned a 1.533 kb PCR product, which included AMN exon 1 to 3. Subsequently, we performed direct Sanger sequencing of 24 independent clones, 12 for each patient. Sequencing analysis revealed 12 clones which contained only the exon 1 frameshift mutation (p.Gln12Argfs*5) and 11 clones showing exclusively the exon 3 missense mutation (p.Met69Lys) indicating inheritance from both parents and confirming the presence of a compound heterozygous mutations. Whereas the frameshift mutation is obviously most likely pathogenic, the missense mutation is predicted to be disease causing by various web-based in silico prediction programs including “Mutation Taster” (http://www.mutationtaster.org/), ‘Sorting Tolerant From Intolerant’ (SIFT) algorithm (http://siftdna.org/), and Panther (http://www.pantherdb.org/). In contrast, the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) program predicts that the amnionless p.Met69Lys missense variant might be a benign amino acid change (score: 0.131).
Additionally, the direct sequencing of the CUBN gene revealed the presence of a heterozygous missense variant p.Val2865Met in exon 52 in both affected half-sisters. This variant (rs146847375) has been reported in the Exome Variant Server (EVS) database (http://evs.gs.washington.edu/EVS/) and is present in 6/4294 (0.14 %) European Americans with a pathogenic polyphen-2 prediction score of 0.996. We did not find any additional potential heterozygous variants in CUBN in our patients. This almost excludes CUBN defects as disease causing in this family, although, we can’t fully exclude the possibility that this heterozygous CUBN variant might contribute to the disease phenotype in our patients with bi-allelic AMN mutations. CUBN is a very large exon-rich gene with more than 200 different missense or truncating variants reported in the EVS database predicted to be possibly or probably protein damaging. Therefore, we reason that the p.Val2865Met missense variant has been an incidental finding. Cases of digenic mutations, where there is one mutant allele in CUBN and one in AMN have to our knowledge never been reported.
Conclusion
Using targeted Sanger sequencing of AMN and CUBN, we identified novel compound heterozygous mutations in AMN in a family from the United Kingdom with typical clinical features of Imerslund-Gräsbeck Syndrome.
Consent
Following informed consent, DNA was obtained from both affected patients. This study was approved by the Northern and Yorkshire Regional Ethics Committee. Patients have given their consent for the Case reports to be published.
Abbreviations
- EVS:
-
Exome Variant Server
- ExAC:
-
Exome Aggregation Consortium
- HGMD:
-
Human Gene Mutation Database
- IGS:
-
Imerslund-Gräsbeck Syndrome
- LMW:
-
Low molecular weight
- SIFT:
-
Sorting Tolerant From Intolerant
References
Imerslund O. Idiopathic chronic megaloblastic anemia in children. Acta Paediatr Suppl. 1960;49 Suppl 119:1–115.
Grasbeck R, Gordin R, Kantero I, Kuhlback B. Selective vitamin B12 malabsorption and proteinuria in young people. A syndrome Acta Med Scand. 1960;167:289–96.
Ovunc B, Otto EA, Vega-Warner V, Saisawat P, Ashraf S, Ramaswami G, et al. Exome sequencing reveals cubilin mutation as a single-gene cause of proteinuria. J Am Soc Nephrol. 2011;22:1815–20.
Aminoff M, Carter JE, Chadwick RB, Johnson C, Grasbeck R, Abdelaal MA, et al. Mutations in CUBN, encoding the intrinsic factor-vitamin B12 receptor, cubilin, cause hereditary megaloblastic anaemia 1. Nat Genet. 1999;21:309–13.
Tanner SM, Aminoff M, Wright FA, Liyanarachchi S, Kuronen M, Saarinen A, et al. Amnionless, essential for mouse gastrulation, is mutated in recessive hereditary megaloblastic anemia. Nat Genet. 2003;33:426–9.
Grasbeck R, Tanner SM. Juvenile selective vitamin B12 malabsorption: 50 years after its description-10 years of genetic testing. Pediatr Res. 2011;70:222–8.
Tanner SM, Li Z, Bisson R, Acar C, Oner C, Oner R, et al. Genetically heterogeneous selective intestinal malabsorption of vitamin B12: founder effects, consanguinity, and high clinical awareness explain aggregations in Scandinavia and the Middle East. Hum Mutat. 2004;23:327–33.
Tanner SM, Sturm AC, Baack EC, Liyanarachchi S, de la Chapelle A. Inherited cobalamin malabsorption. Mutations in three genes reveal functional and ethnic patterns Orphanet J Rare Dis. 2012;7:56.
Kalantry S, Manning S, Haub O, Tomihara-Newberger C, Lee HG, Fangman J, et al. The amnionless gene, essential for mouse gastrulation, encodes a visceral-endoderm-specific protein with an extracellular cysteine-rich domain. Nat Genet. 2001;27:412–6.
Fyfe JC, Madsen M, Hojrup P, Christensen EI, Tanner SM, de la Chapelle A, et al. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood. 2004;103:1573–9.
Zhai XY, Nielsen R, Birn H, Drumm K, Mildenberger S, Freudinger R, et al. Cubilin- and megalin-mediated uptake of albumin in cultured proximal tubule cells of opossum kidney. Kidney Int. 2000;58:1523–33.
Tanner SM, Li Z, Perko JD, Oner C, Cetin M, Altay C, et al. Hereditary juvenile cobalamin deficiency caused by mutations in the intrinsic factor gene. Proc Natl Acad Sci U S A. 2005;102:4130–3.
Dickson LE, Wagner MC, Sandoval RM, Molitoris BA. The proximal tubule and albuminuria: really! J Am Soc Nephrol. 2014;25:443–53.
Verroust PA, Christensen EA. Megalin and cubilin–the story of two multipurpose receptors. Neph Dial Transpl. 2002;17:1867–71.
Amsellem S, Gburek J, Hamard G, Nielsen R, Willnow TE, Devuyst O, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21:1859–67.
Wahlstedt-Froberg V, Pettersson T, Aminoff M, Dugue B, Grasbeck R. Proteinuria in cubilin-deficient patients with selective vitamin B12 malabsorption. Pediatr Nephrol. 2003;18:417–21.
Levin-Iaina N, Dinour D, Morduchowicz G, Ganon L, Holtzman EJ. Molecular study of proteinuria in patients treated with B12 supplements: do not forget megaloblastic anemia type 1. Nephron Clin Pract. 2011;118:c67–71.
Acknowledgements
We acknowledge generous support from the Northern Counties Kidney Research Fund (http://www.nckrf.org.uk/).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EM drafted the manuscript. AMH, VVW and EAO carried out the molecular genetic studies and drafted the manuscript, LB, SJ and JAG contributed clinical data and revised it critically for important intellectual content. JAS conceived of the study, and participated in its design and coordination and helped to draft and revise the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Montgomery, E., Sayer, J.A., Baines, L.A. et al. Novel compound heterozygous mutations in AMN cause Imerslund-Gräsbeck syndrome in two half-sisters: a case report. BMC Med Genet 16, 35 (2015). https://doi.org/10.1186/s12881-015-0181-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-015-0181-2