
Abstract
Background
Early detection of Zika virus (ZIKV) infection during the viremia and viruria facilitates proper patient management and mosquito control measurement to prevent disease spread. Therefore, a cost-effective nucleic acid detection method for the diagnosis of ZIKV infection, especially in resource-deficient settings, is highly required.
Methods
In the present study, a single-tube reverse transcription loop-mediated isothermal amplification (RT-LAMP) assay was developed for the detection of both the Asian and African-lineage ZIKV. The detection limit, strain coverage and cross-reactivity of the ZIKV RT-LAMP assay was evaluated. The sensitivity and specificity of the RT-LAMP were also evaluated using a total of 24 simulated clinical samples. The ZIKV quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assay was used as the reference assay.
Results
The detection limit of the RT-LAMP assay was 3.73 ZIKV RNA copies (probit analysis, P ≤ 0.05). The RT-LAMP assay detected the ZIKV genomes of both the Asian and African lineages without cross-reacting with other arthropod-borne viruses. The sensitivity and specificity of the RT-LAMP assay were 90% (95% CI = 59.6–98.2) and 100% (95% CI = 78.5–100.0), respectively. The RT-LAMP assay detected ZIKV genome in 9 of 24 (37.5%) of the simulated clinical samples compared to 10 of 24 (41.7%) by qRT-PCR assay with a high level of concordance (κ = 0.913, P < 0.001).
Conclusion
The RT-LAMP assay is applicable for the broad coverage detection of both the Asian and African ZIKV strains in resource-deficient settings.
Similar content being viewed by others


Background
The Aedes mosquito-borne Zika virus (ZIKV) was discovered in Uganda in 1947 [1] and until recently, caused only sporadic human infections in Africa and Asia characterized by mild and self-limiting disease [2,3,4]. An epidemic affecting almost three quarters of the population in Yap Island, Federated States of Micronesia in 2007 was the first outbreak of ZIKV outside of Africa and Asia [5]. Following that, in 2013–2014, ZIKV rapidly spread to other countries in the Pacific, including French Polynesia [6], New Caledonia [7], the Cook Islands, Solomon Islands and Easter Island [8, 9]. Notably, the French Polynesia outbreak resulted in almost 9000 suspected cases, with an unusual increase of incidences involving neurological deformities among infants born after the outbreak [10]. In early 2015, ZIKV was identified in an outbreak in Bahia, Brazil [11] and by November 2016, transmission of ZIKV has since been reported in at least 48 other countries in the Americas [12]. It was during the Brazil outbreak that health authorities observed a surge in adverse pregnancy outcomes including congenital microcephaly, cerebral calcification and fetal growth restriction, among infants born to ZIKV-infected mothers [13]. Adults were clinically diagnosed with Guillain-Barré syndrome following ZIKV infection [8, 14], although the associated pathophysiological mechanisms have yet to be ascertained. The possible associations of ZIKV infection to these health disorders compelled the World Health Organization to declare ZIKV a Public Health Emergency of International Concern on February 2016 [15].
The ZIKV epidemics highlighted the unfortunate circumstances that this disease undeservedly affected most the economically marginalized populations [16] and by chance, they happened to be living in regions where Aedes mosquitoes thrive [17]. The expansion and spread of ZIKV, however, can be attributed to the wider geographical reach of Ae. albopictus that unlike Ae. aegypti can survive in temperate areas [17]. In addition, the infection can be transmitted through sexual contact [18, 19], mother-to-fetus [20,21,22] and via blood transfusion [23,24,25], albeit only in rare instances. Considering its far reaching impact of the infection, it is crucial that the development of rapid, cost-effective assays and portable ZIKV detection instruments to be expedited for deployment to the most needing.
Current ZIKV diagnosis use either serological assays detecting ZIKV-reactive antibodies or molecular detection of ZIKV RNA [26]. Serological assays detecting specific anti-ZIKV antibodies and the neutralization tests detecting specific ZIKV neutralizing antibodies are laborious, time-consuming and complicate by possible cross-reactivity with other flaviviruses [26, 27]. ZIKV RNA detection by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) [28, 29] is often considered the gold standard as it provides definitive diagnosis. This method however, requires usage of costly reagents and amplification and detection equipment. The method also requires trained laboratory personnel to perform. The loop-mediated isothermal amplification (LAMP) method that provides simple, sensitive and rapid nucleic acid amplification under isothermal conditions promises to be a good alternative to qRT-PCR [30]. The method has been used for the detection of various RNA viruses including ZIKV [31,32,33,34,35,36,37,38,39,40,41,42,43]. Majority of the earlier reports of RT-LAMP for ZIKV used primers designed based on the lineage-specific regions of ZIKV genome [31,32,33,34,35,36,37,38,39,40]. In the present study, we described an improved RT-LAMP assay which took into account the detection of ZIKV strains from both the African and Asian lineages. The ZIKV RT-LAMP assay described here was evaluated for ZIKV RNA detection from simulated serum, saliva, and urine specimens.
Methods
Zika viruses
Three Asian ZIKV strains (P6–740, PRVABC59 and H/PF/2013) [44,45,46] and one African ZIKV strain (MR766) [1] were used. Strain P6–740 was obtained from Dr. Robert Tesh (World Reference Center for Emerging Viruses and Arboviruses, The University of Texas Medical Branch, Galveston, USA). Strain PRVABC59 was maintained in the laboratory by K. Zandi (author). Strain H/PF/2013 and MR766 were provided by Dr. Li-Sze Lim (Medical Innovation Ventures Pte. Ltd., Malaysia). The viruses were propagated in Vero cells using Dulbecco’s modified Eagle medium (DMEM, Gibco, NY, USA) supplemented with 2% heat-inactivated fetal bovine serum (FBS collected in South America, Capricorn, Germany), 0.1 mM non-essential amino acids (NEAA) and 2 mM L-glutamine. The infected cells were incubated at 37 °C in 5% CO2 atmosphere for 7 days and the infected cell culture supernatants were then harvested and stored at − 80 °C until further use.
Simulated clinical samples
The study was approved by the UM Institutional Biosafety and Biosecurity Committee (Approval Number: UMIBBC/NOI/R/TNCPNI/TIDREC-007/22072020) and the UMMC Medical Ethics Committee (Ethics Committee/IRB Reference Number: 908.11). All ZIKV-positive simulated clinical samples were prepared by spiking the viral culture supernatant into actual human saliva, urine and serum. The viral culture supernatant was serially diluted with the serum-free media to viral titers ranging from 10− 2 to 104 plaque-forming unit/ml (PFU/ml). The diluted viral culture supernatants were then mixed with the saliva, urine and serum samples at a 1:9 ratio. The final viral titers of the simulated clinical samples ranged from 10− 3 to 103 PFU/ml. The human saliva, urine and serum without spiked ZIKV were used as ZIKV-negative simulated clinical samples. A total of 24 simulated clinical samples consisting 8 saliva, 8 urine and 8 serum samples were prepared (Additional file 1: Table S1). The saliva and serum samples were obtained from the same healthy donor, while the urine sample was obtained from another healthy donor. Only Asian ZIKV strains P6–740 was used for the preparation of simulated clinical samples.
Viral plaque assay
The viral plaque assay was performed to determine the infectious titer of the ZIKV used for the preparation of simulated clinical samples. Briefly, the Vero cells were seeded at a density of 2 × 105 cells/well in 24-wells plate and allowed to grow overnight until more than 80% confluency in DMEM supplemented with 10% FBS, 0.1 mM NEAA and 2 mM L-glutamine. The overnight culture media was replaced with a 10-fold serially diluted virus stock in the serum-free media (200 μl of each virus dilution). The plate was placed on a rocker and gently rocked for 1 h for virus adsorption. Subsequently, the virus supernatants were removed from each well and overlaid with 1 ml of DMEM supplemented with 2% FBS, 0.8% carboxymethylcellulose (CMC, Sigma-Aldrich, USA), 0.1 mM NEAA and 2 mM L-glutamine. The plate was incubated at 37 °C in 5% CO2 atmosphere for 5 days. After 5 days of incubation, the overlaid media was removed and cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, USA) for 30 min at room temperature. The cells were then washed using 1 × PBS for three times. Finally, 0.5% crystal violet in 20% ethanol (Sigma-Aldrich, USA) was used to stain the cells for 15 min in order to enable the visibility of virus plaques. The virus plaques were counted using the SMZ 1000 stereomicroscope (Nikon, Tokyo, Japan) and the virus infectious titer was expressed in PFU/ml.
RNA extraction
The infected cell culture supernatant and simulated clinical serum samples (140 μl) were subjected to RNA extraction using QIAamp Viral RNA Mini Kit (Qiagen, Germany) following the manufacturer’s protocol. The eluted RNA (60 μl) was stored at − 80 °C until further use.
Design of ZIKV-specific RT-LAMP assay primers
The virus genomes of both Asian and African lineages were retrieved from GenBank. The genomes of other flaviviruses were also included for comparison. The sequences were aligned using Clustal X 2.0 [47]. The RT-LAMP primers were designed following the criteria previously described [30]. Conserved regions among flaviviruses were identified and excluded from the primer sequences. The RT-LAMP primers designed were exhaustically compared with an alignment of ZIKV genomes retrieved from GenBank. The five nucleotides at the 3′ end of F3, B3, FLP and BLP primers as well as the five nucleotides at both 3′ and 5′ end of FIP and BIP primers were considered critical sites for priming and amplification. The coverage of the primers was further validated by assessing the assay using both Asian and African ZIKV strains.
RT-LAMP assay
The RT-LAMP reaction was prepared using the Loopamp RNA Amplification Kit (Eiken Chemical Co. Ltd., Japan). Each RT-LAMP reaction (25 μl) was added with the inner primers (20 pmol each), outer primers (2.5 pmol each), loop primer (20 pmol each), Fluorescent Detection Reagent (Eiken Chemical Co. Ltd., Japan; 1 μl), and the eluted RNA (5 μl). The RT-LAMP were performed using LA-500 Loopamp real-time turbidimeter (Eiken Chemical Co. Ltd., Japan) according to the following conditions: 90 min at 63 °C followed by 5 min of assay inactivation at 80 °C. The turbidity of RT-LAMP reaction was measured at 650 nm every 6 s. The threshold time (Tt) value was recorded when the turbidity crossed the threshold cut-off value at 0.07 absorbance units [48, 49].
Cross-reactivity of RT-LAMP assay
The cross-reactivity of the ZIKV RT-LAMP primers was evaluated against other arboviruses including dengue virus type 1 (DENV-1), DENV-2, DENV-3, DENV-4, Japanese encephalitis virus (JEV), Langat virus (LGTV), Sindbis virus (SINV), Chikungunya virus (CHIKV), and Getah virus (GETV). All these viruses were attained from the TIDREC viral repository [48,49,50,51,52]. Nuclease-free water was used as the negative control.
Detection limit of RT-LAMP assay
The detection limit of the RT-LAMP assay was assessed by using a 10-fold serially diluted ZIKV RNA (ranged from 1 to 103 RNA copies) extracted from the virus culture supernatant. The initial ZIKV RNA copy number in the culture supernatant stock used were quantitated using the qRT-PCR assay. The detection limit test of RT-LAMP was repeated four times. Nuclease-free water was used as the negative control and as the diluent for the preparation of serially diluted viral RNA.
Evaluation of RT-LAMP assay
The feasibility of the RT-LAMP assay for detection of ZIKV RNA in clinical setting was assessed using the simulated clinical samples as described above. The qRT-PCR was used as a reference assay for the detection of ZIKV RNA in the samples. Results from the RT-LAMP and qRT-PCR assays were compared.
Real-time qRT-PCR assay (reference assay)
The viral RNA and samples used were quantitated using the Genesig Real-Time qRT-PCR ZIKV Detection Kit (PrimerDesign Ltd., UK). The Genesig qRT-PCR assay standard plot was prepared using a 10-fold serially diluted synthetic ZIKV RNA template with known copy number (ranged from 10 to 106 RNA copies). The qRT-PCR reaction consists of the real time master mix (10 μl), probe/primer mix (1 μl), nuclease-free water (4 μ), and the extracted RNA (5 μl), in a 20 μl final reaction volume. The qRT-PCR were performed according to the following conditions: 10 min at 55 °C, 8 min at 95 °C followed by 50 cycles of 10 s at 95 °C and 60 s at 60 °C using StepOnePlus real time PCR system (Applied Biosystems, USA). The threshold cycles (Ct) obtained were used to determine the ZIKV RNA copy number in the samples based on standard curve from qRT-PCR using StepOne Software v2.2.1.
Statistical analysis
A probit analysis (P ≤ 0.05) was performed to determine the detection limit of the RT-LAMP assay. The degree of agreement [kappa value (κ), P < 0.001] between RT-LAMP and qRT-PCR test results was measured. The degree of agreement and probit analyses were performed using IBM SPSS Statistics, version 21 (IBM Corporation, New York, United States). The diagnostic performance of RT-LAMP compared to qRT-PCR was determined using web-based EBM Diagnostic Test Calculator (http://ebm-tools.knowledgetranslation.net/calculator/diagnostic/).
Results
Design of ZIKV-specific RT-LAMP assay primers
A set of six RT-LAMP primers comprising two outer, two inner, and two loop primers was designed (Table 1 and Additional file 2: Fig. S1) to target both Asian and African ZIKV strains. This was accomplished using the alignment of the conserved NS2A-NS3 junction of the ZIKV genome; each lineage-specific consensus was derived from at least five ZIKV strains (Additional file 3: Table S2). The sequences of the RT-LAMP primers were further compared with an alignment of 463 ZIKV genomes retrieved from GenBank (Additional file 4: Figure S2). In this comparison, BIP and FLP primers showed no critical nucleotide mismatch with all ZIKV genomes. Whereas the F3, B3, FIP and BLP primers respectively showed 0.22% (1/463), 1.94% (9/463), 1.30% (6/463) and 1.30% (6/463) of critical nucleotide mismatches with the ZIKV genomes. Figure 1 showed that the RT-LAMP primers detected the four reference ZIKV strains.

Amplification curves of the ZIKV RT-LAMP assay. A-D, four ZIKV strains; E-M, four DENV, JEV, LGTV, SINV, CHIKV and GETV, respectively; N, negative control; O, y- and x-axis titles
Cross-reactivity of RT-LAMP assay
No cross-reactivity of the RT-LAMP assay was observed with all other arthropod-borne viruses including all four DENV serotypes, JEV, LGTV, SINV, CHIKV and GETV (Fig. 1).
Detection limit of RT-LAMP assay
The detection limit of the RT-LAMP assay was assessed by repeatedly testing on the serially diluted ZIKV RNA with known copy number (Fig. 2 and Additional file 5: Table S3). The positive detection by RT-LAMP assays (n = 4) for 1000, 100, 10, and 1 ZIKV RNA copy were 100% (4 of 4), 100% (4 of 4), 100% (4 of 4), and 25% (1 of 4), respectively, with the mean time threshold (Tt) of 26.95 ± 1.25 min, 29.70 ± 16.14 min, 44.18 ± 12.37 min, 39.10 min, respectively. The detection limit of the RT-LAMP assay was 3.73 ZIKV RNA copies (probit analysis, P ≤ 0.05) (Fig. 3).

Time threshold of positivity for RT-LAMP assays of serially diluted ZIKV RNA. The mean of Tt-values was calculated with available positive results out of four replicates. Error bars indicate the standard deviations of Tt-values from the mean

Detection limit of the ZIKV RT-LAMP assay. The probit regression curve was obtained from four replicates of ZIKV RNA in four dilutions (1000, 100, 10, and 1 copy numbers)
Evaluation of RT-LAMP assay
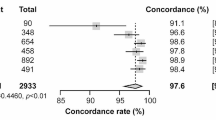
The RT-LAMP assay for detection of ZIKV RNA was assessed by testing on a total of 24 simulated clinical samples. No evaluation was performed on actual clinical samples as none are currently available at the center. To date no Zika patient has been seen in our hospital. The diagnostic performance of the RT-LAMP in comparison with qRT-PCR assay was summarized in Table 2. The sensitivity and specificity of the RT-LAMP assay were 90% (95% CI = 59.6–98.2) and 100% (95% CI = 78.5–100.0), respectively. The RT-LAMP assay detected ZIKV genome in 9 of 24 (37.5%) of the samples compared to 10 of 24 (41.7%) by qRT-PCR assay, with a high concordance of κ = 0.913 (P < 0.001) between these two methods. ZIKV RNA was tested negative in one urine sample by RT-LAMP but positive by qRT-PCR; this sample contained 3.02 log10 copies of RNA/ml or 1 PFU/ml (Fig. 4). Samples containing 10–103 PFU/ml (n = 9) were tested positive by both RT-LAMP and qRT-PCR, whereas samples containing 10− 1–10− 3 PFU/ml (n = 9) and the negative samples without spiked ZIKV (n = 3) were tested negative by both RT-LAMP and qRT-PCR.

ZIKV RNA copy numbers of the simulated clinical human saliva, urine and serum specimens that tested positive by qRT-PCR according to the infectious virus titer (n = 10). The dashed line indicates the detection limit of qRT-PCR. The error bars indicate the standard deviation of the viral RNA copy numbers from the mean. Open circle, positive by qRT-PCR only; filled circle, positive by both qRT-PCR and RT-LAMP
Discussion
In the present study, a RT-LAMP assay was developed for isothermal detection of ZIKV RNA in simulated clinical specimens. The RT-LAMP assay could detect all the ZIKV strains used in this study without cross-reacting with a number of other common arthropod-borne viruses including all four DENV serotypes, JEV, LGTV, SINV, CHIKV and GETV. The RT-LAMP assay was sensitive, specific and useful for broad coverage detection of both the Asian and African ZIKV lineages.
Recently, several research groups have developed RT-LAMP assays for the detection of ZIKV by targeting the different regions of the ZIKV genome with excellent sensitivity and specificity [31,32,33,34,35,36,37,38,39,40,41,42,43]. They however, designed the primers for the RT-LAMP assays based only on a single lineage of ZIKV, which is either Asian [33,34,35,36,37, 39, 40] or African [32] lineage. Many studies designed the RT-LAMP primers based primarily on the sequences of the Asian ZIKV since the recent epidemics in the Pacific islands and Americas [5,6,7, 11] were caused by ZIKV from the Asian lineage [53]. Nonetheless, the possibility of re-emergence of the African lineage should not be excluded. Chotiwan et al. (2017) suggested using two sets of RT-LAMP primers, one for Asian ZIKV and another one for the African ZIKV [31]. The two-tube RT-LAMP assay, however, would definitely increase the cost of diagnosis. On the other hand, Kurosaki et al. (2017) combined two sets of RT-LAMP primers (total 11 primers) in single tube for simultaneous detection of Asian and African ZIKV [41]. Nonetheless, the use of more than six primers is often not preferable in RT-LAMP assay to avoid false positive due to a primer self-amplification [54]. Recently, Escalante-Maldonado et al. (2019) and Bui et al. (2020) developed RT-LAMP assay using a set of six primers based on the 64 and 130 ZIKV sequences retrieved from GenBank, respectively [42, 43]. Here, we designed a set of six RT-LAMP primers without critical mismatches with at least 95% of the 463 ZIKV genome sequences for broad coverage detection of both the Asian and African ZIKV lineages in a single-tube assay.
The detection limit of the RT-LAMP assay (~ 4 copies of ZIKV RNA) was comparable to that of ZIKV RT-LAMP assays previously reported; the detection limits ranged from 1 to 111 copies of ZIKV RNA [31, 32, 35]. In this study, the ZIKV RNA copy/infectious particle ratio in the simulated clinical specimens ranged from 102 to 103. Similar ZIKV RNA copy/infectious particle ratios have also been previously reported [55, 56]. Thus, the RT-LAMP assay is a potentially useful diagnostic tool for detecting viremic patients who are potentially contagious. Early detection of the viremic patients would permit immediate execution of proper disease control measures such as deploying insecticidal spraying and activating community-based mosquito control activities. Early diagnosis would also help clinicians in providing proper supportive treatment and counselling of Zika patients without needing to prescribe unnecessary medications.
Zika virus has been detected in human blood, saliva [57, 58], urine [59,60,61], semen [62, 63] and breastmilk [64]. Among all types of specimens, blood, saliva and urine are the common specimens used for Zika diagnosis. It has been reported that the viral titers in the patient’s blood, during the acute infection, ranged from 103 to 106 RNA copies/ml [65]. The viremic phase of ZIKV infection, however, is short with less than 2 to 5 days after fever onset [57, 59, 60]. The use of saliva has been shown to improve the detection of ZIKV RNA but did not enlarge the window of detection [57]. In contrast, shedding of ZIKV in urine persists for ~ 2 weeks [60] and sometimes it can be up to > 20 days after fever onset [59]. The use of urine specimen, therefore, is a practical way to enlarge the window of ZIKV detection. Here, we demonstrated that the RT-LAMP assay detected the simulated clinical specimens (serum, saliva, and urine) with viral load of as low as 10 PFU/ml, which is equivalent to approximately 103 to 104 RNA copies/ml. This detection sensitivity would be sufficient to diagnose viruric patients if their urine samples are collected within 10 days after fever onset considering viruric patients usually has viral load of > 103 RNA copies/ml during the first 10 days of ZIKV infection [59].
There are, however, several limitations to our study. The performance of the RT-LAMP assay has not been validated on any real clinical samples. The evaluation was done in a very small cohort of simulated clinical samples (n = 24), in which the virus titers could be controlled. Therefore, there is no assurance of similar performance in field diagnostic settings.
Conclusions
The RT-LAMP assay developed in this study was specific and sensitive for broad coverage detection of both the Asian and African ZIKV strains. The RT-LAMP assay can greatly enhance diagnosis of Zika in situation where resources are deficient.
Availability of data and materials
All data generated or analyzed in this study are included in this published article and its Additional files.
Abbreviations
- Ae.:
-
Aedes
- B3:
-
Backward outer primer
- BIP:
-
Backward inner primer
- BLP:
-
Backward loop primer
- CHIKV:
-
Chikungunya virus
- CI:
-
Confidence interval
- CMC:
-
Carboxymethylcellulose
- CO2 :
-
Carbon dioxide
- DENV:
-
Dengue virus
- DMEM:
-
Dulbecco’s modified Eagle medium
- F3:
-
Forward outer primer
- FBS:
-
Fetal bovine serum
- FIP:
-
Forward inner primer
- FLP:
-
Forward loop primer
- GETV:
-
Getah virus
- IAS:
-
Institute for Advanced Studies
- JEV:
-
Japanese encephalitis virus
- LGTV:
-
Langat virus
- NEAA:
-
Non-essential amino acids
- NS:
-
Non-structural
- PBS:
-
Phosphate-buffered saline
- PFU:
-
Plaque-forming unit
- qRT-PCR:
-
Quantitative reverse transcription-polymerase chain reaction
- RNA:
-
Ribonucleic acid
- RT-LAMP:
-
Reverse transcription loop-mediated isothermal amplification
- SINV:
-
Sindbis virus
- TIDREC:
-
Tropical Infectious Diseases Research and Education Centre
- Tt:
-
Time threshold
- UM:
-
Universiti Malaya
- UMMC:
-
Universiti Malaya Medical Centre
- ZIKV:
-
Zika virus
References
Dick GW, Kitchen SF, Haddow AJ. Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg. 1952;46(5):509–20.
Fagbami AH. Zika virus infections in Nigeria: virological and seroepidemiological investigations in Oyo state. J Hyg (Lond). 1979;83(2):213–9.
Macnamara FN. Zika virus: a report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans R Soc Trop Med Hyg. 1954;48(2):139–45.
Olson JG, Ksiazek TG, Suhandiman, Triwibowo. Zika virus, a cause of fever in Central Java, Indonesia. Trans R Soc Trop Med Hyg. 1981; 75 (3): 389–93.
Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med. 2009;360(24):2536–43.
Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, Sall AA, Musso D. Zika virus, French polynesia, south pacific, 2013. Emerg Infect Dis. 2014;20(6):1085–6.
Dupont-Rouzeyrol M, O'Connor O, Calvez E, Daures M, John M, Grangeon JP, Gourinat AC. Co-infection with Zika and dengue viruses in 2 patients, New Caledonia, 2014. Emerg Infect Dis. 2015;21(2):381–2.
Musso D, Nilles EJ, Cao-Lormeau VM. Rapid spread of emerging Zika virus in the Pacific area. Clin Microbiol Infect. 2014;20(10):O595–6.
Higgs S, Vanlandingham DL. Search and one will find: Zika virus everywhere. Trans R Soc Trop Med Hyg. 2016;110(4):207–8.
Besnard M, Eyrolle-Guignot D, Guillemette-Artur P, Lastere S, Bost-Bezeaud F, Marcelis L, Abadie V, Garel C, Moutard ML, Jouannic JM, et al. Congenital cerebral malformations and dysfunction in fetuses and newborns following the 2013 to 2014 Zika virus epidemic in French Polynesia. Euro Surveill. 2016;21(13). https://doi.org/10.2807/1560-7917.ES.2016.21.13.30181.
Campos GS, Bandeira AC, Sar di S I. Zika virus outbreak, Bahia, Brazil. Emerg Infect Dis. 2015;21(10):1885–6.
WHO: Zika virus microcephaly and Guillain-Barré syndrome Situation Report. 2016 http://apps.who.int/iris/bitstream/10665/251648/1/zikasitrep24Nov16-eng.pdf?ua=1.
Brasil P, Pereira JP Jr, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, Rabello RS, Valderramos SG, Halai UA, Salles TS, et al. Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med. 2016; 375 (24): 2321–34.
Langerak T, Yang H, Baptista M, Doornekamp L, Kerkman T, Codrington J, Roosblad J, Vreden SG, De Bruin E, Mogling R, et al. Zika virus infection and Guillain-Barre syndrome in three patients from Suriname. Front Neurol. 2016; 7: 233.
WHO: WHO statement on the first meeting of the International Health Regulations (2005) (IHR 2005) Emergency Committee on Zika virus and observed increase in neurological disorders and neonatal malformations. 2015. http://www.who.int/mediacentre/news/statements/2016/1st-emergency-committee-zika/en/.
Hotez PJ, Bottazzi ME, Strych U, Chang LY, Lim YA, Goodenow MM, AbuBakar S. Neglected tropical diseases among the Association of Southeast Asian Nations (ASEAN): overview and update. PLoS Negl Trop Dis. 2015;9(4):e0003575.
Sakkas H, Economou V, Papadopoulou C. Zika virus infection: past and present of another emerging vector-borne disease. J Vector Borne Dis. 2016;53(4):305–11.
Foy BD, Kobylinski KC, Chilson Foy JL, Blitvich BJ, Travassos da Rosa A, Haddow AD, Lanciotti RS, Tesh RB. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis. 2011; 17 (5): 880-2.
Musso D, Roche C, Robin E, Nhan T, Teissier A, Cao-Lormeau VM. Potential sexual transmission of Zika virus. Emerg Infect Dis. 2015;21(2):359–61.
Besnard M, Lastere S, Teissier A, Cao-Lormeau V, Musso D. Evidence of perinatal transmission of Zika virus, French Polynesia, December 2013 and February 2014. Euro Surveill. 2014;19(13):20751.
Oliveira Melo AS, Malinger G, Ximenes R, Szejnfeld PO, Alves Sampaio S, Bispo de Filippis AM. Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound Obstet Gynecol. 2016;47(1):6–7.
Oliveira DB, Almeida FJ, Durigon EL, Mendes EA, Braconi CT, Marchetti I, Andreata-Santos R, Cunha MP, Alves RP, Pereira LR, et al. Prolonged shedding of Zika virus associated with congenital infection. N Engl J Med. 2016;375(12):1202–4.
Musso D, Nhan T, Robin E, Roche C, Bierlaire D, Zisou K, Shan Yan A, Cao-Lormeau VM, Broult J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Euro Surveill. 2014;19(14):20761.
Gallian P, Cabie A, Richard P, Paturel L, Charrel RN, Pastorino B, Leparc-Goffart I, Tiberghien P, de Lamballerie X. Zika virus in asymptomatic blood donors in Martinique. Blood. 2017;129(2):263–6.
Motta IJ, Spencer BR, Cordeiro da Silva SG, Arruda MB, Dobbin JA, Gonzaga YB, Arcuri IP, Tavares RC, Atta EH, Fernandes RF, et al. Evidence for transmission of Zika virus by platelet transfusion. N Engl J Med. 2016;375(11):1101–3.
Waggoner JJ, Pinsky BA. Zika virus: diagnostics for an emerging pandemic threat. J Clin Microbiol. 2016;54(4):860–7.
Charrel RN, Leparc-Goffart I, Pas S, de Lamballerie X, Koopmans M, Reusken C. Background review for diagnostic test development for Zika virus infection. Bull World Health Organ. 2016;94(8):574–584D.
Faye O, Faye O, Diallo D, Diallo M, Weidmann M, Sall AA. Quantitative real-time PCR detection of Zika virus and evaluation with field-caught mosquitoes. Virol J. 2013;10:311.
Yang Y, Wong G, Ye B, Li S, Li S, Zheng H, Wang Q, Liang M, Gao GF, Liu L, et al. Development of a reverse transcription quantitative polymerase chain reaction-based assay for broad coverage detection of African and Asian Zika virus lineages. Virol Sin. 2017;32(3):199–206.
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):E63.
Chotiwan N, Brewster CD, Magalhaes T, Weger-Lucarelli J, Duggal NK, Ruckert C, Nguyen C, Garcia Luna SM, Fauver JR, Andre B, et al. Rapid and specific detection of Asian- and African-lineage Zika viruses. Sci Transl Med. 2017;9(388):eaag0538.
Lee D, Shin Y, Chung S, Hwang KS, Yoon DS, Lee JH. Simple and highly sensitive molecular diagnosis of Zika virus by lateral flow assays. Anal Chem. 2016;88(24):12272–8.
Song J, Mauk MG, Hackett BA, Cherry S, Bau HH, Liu C. Instrument-free point-of-care molecular detection of Zika virus. Anal Chem. 2016;88(14):7289–94.
Tian B, Qiu Z, Ma J, Zardan Gomez de la Torre T, Johansson C, Svedlindh P, Stromberg M. Attomolar Zika virus oligonucleotide detection based on loop-mediated isothermal amplification and AC susceptometry. Biosens Bioelectron. 2016;86:420–5.
Wang X, Yin F, Bi Y, Cheng G, Li J, Hou L, Li Y, Yang B, Liu W, Yang L. Rapid and sensitive detection of Zika virus by reverse transcription loop-mediated isothermal amplification. J Virol Methods. 2016;238:86–93.
Calvert AE, Biggerstaff BJ, Tanner NA, Lauterbach M, Lanciotti RS. Rapid colorimetric detection of Zika virus from serum and urine specimens by reverse transcription loop-mediated isothermal amplification (RT-LAMP). PLoS One. 2017;12(9):e0185340.
Sabalza M, Yasmin R, Barber CA, Castro T, Malamud D, Kim BJ, Zhu H, Montagna RA, Abrams WR. Detection of Zika virus using reverse-transcription LAMP coupled with reverse dot blot analysis in saliva. PLoS One. 2018;13(2):e0192398.
Lamb LE, Bartolone SN, Tree MO, Conway MJ, Rossignol J, Smith CP, Chancellor MB. Rapid detection of Zika virus in urine samples and infected mosquitos by reverse transcription-loop-mediated isothermal amplification. Sci Rep. 2018;8(1):3803.
Zhao J, Feng R. Sensitive and rapid detection of Zika virus by loop-mediated isothermal amplification. Virus Genes. 2019;55(1):43–50.
Lin X, Wu M, Wang W, Gao Y, Zhang W, Wu D, Wu Y, Zhou X, Li G. Visual detection of Zika virus by isothermal nucleic acid amplification combined with a lateral-flow device. Anal Methods. 2019;11:1795–801.
Kurosaki Y, Martins DBG, Kimura M, Catena ADS, Borba M, Mattos SDS, Abe H, Yoshikawa R, de Lima Filho JL, Yasuda J. Development and evaluation of a rapid molecular diagnostic test for Zika virus infection by reverse transcription loop-mediated isothermal amplification. Sci Rep. 2017;7(1):13503.
Escalante-Maldonado O, Gavilán RG, García MP, Marcelo A, Pacheco E, Cabezas C, Yamazaki W. Development and validation of loop-mediated isothermal amplification for the detection of the Zika virus. Rev Peru Med Exp Public Health. 2019;36(3):442–7.
Bui TT, Moi ML, Morita K, Hasebe F. Development of universal and lineage-specific primer sets for rapid detection of the Zika virus (ZIKV) in blood and urine samples using one-step reverse transcription loop-mediated isothermal amplification (RT-LAMP). Jpn J Infect Dis. 2020;73(2):153–6.
Marchette NJ, Garcia R, Rudnick A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am J Trop Med Hyg. 1969;18(3):411–5.
Lanciotti RS, Lambert AJ, Holodniy M, Saavedra S, Signor Ldel C. Phylogeny of Zika virus in Western hemisphere, 2015. Emerg Infect Dis. 2016;22(5):933–5.
Baronti C, Piorkowski G, Charrel RN, Boubis L, Leparc-Goffart I, de Lamballerie X. Complete coding sequence of zika virus from a French Polynesia outbreak in 2013. Genome Announc. 2014;2(3):e00500–14.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–82.
Lau YL, Lai MY, Teoh BT, Abd-Jamil J, Johari J, Sam SS, Tan KK, AbuBakar S. Colorimetric detection of dengue by single tube reverse-transcription-loop-mediated isothermal amplification. PLoS One. 2015;10(9):e0138694.
Teoh BT, Sam SS, Tan KK, Johari J, Danlami MB, Hooi PS, Md-Esa R, AbuBakar S. Detection of dengue viruses using reverse transcription-loop-mediated isothermal amplification. BMC Infect Dis. 2013;13:387.
Sam SS, Teoh BT, Chee CM, Mohamed-Romai-Noor NA, Abd-Jamil J, Loong SK, Khor CS, Tan KK, AbuBakar S. A quantitative reverse transcription-polymerase chain reaction for detection of Getah virus. Sci Rep. 2018;8(1):17632.
Muhd Radzi SF, Rückert C, Sam SS, Teoh BT, Jee PF, Phoon WH, AbuBakar S, Zandi K. Detection of Langat virus by TaqMan real-time one-step qRT-PCR method. Sci Rep. 2015;5:14007.
Teoh BT, Sam SS, Tan KK, Danlami MB, Shu MH, Johari J, Hooi PS, Brooks D, Piepenburg O, Nentwich O, Wilder-Smith A, Franco L, Tenorio A, AbuBakar S. Early detection of dengue virus by use of reverse transcription-recombinase polymerase amplification. J Clin Microbiol. 2015;53(3):830–7.
Zanluca C, Melo VC, Mosimann AL, Santos GI, Santos CN, Luz K. First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz. 2015;110(4):569–72.
Senarath KD, Usgodaarachchi RB, Navaratne V, Nagahawatte A, Wijayarathna CD, Alvitigala J, Goonasekara CL. Non specific amplification with the LAMP technique in the diagnosis of tuberculosis in Sri Lankan settings. J Tubercul Res. 2014: 2: 168–72.
Duggal NK, Ritter JM, Pestorius SE, Zaki SR, Davis BS, Chang GJ, Bowen RA, Brault AC. Frequent Zika virus sexual transmission and prolonged viral rna shedding in an immunodeficient mouse model. Cell Rep. 2017;18(7):1751–60.
Xie X, Yang Y, Muruato AE, Zou J, Shan C, Nunes BT, Medeiros DB, Vasconcelos PF, Weaver SC, Rossi SL, et al. Understanding Zika virus stability and developing a chimeric vaccine through functional analysis. MBio. 2017;8(1):e02134–16.
Musso D, Roche C, Nhan TX, Robin E, Teissier A, Cao-Lormeau VM. Detection of Zika virus in saliva. J Clin Virol. 2015;68:53–5.
Barzon L, Pacenti M, Berto A, Sinigaglia A, Franchin E, Lavezzo E, Brugnaro P, Palu G. Isolation of infectious Zika virus from saliva and prolonged viral RNA shedding in a traveller returning from the Dominican Republic to Italy, January 2016. Euro Surveill. 2016;21(10):30159.
Gourinat AC, O'Connor O, Calvez E, Goarant C, Dupont-Rouzeyrol M. Detection of Zika virus in urine. Emerg Infect Dis. 2015; 21 (1): 84–6.
Zhang FC, Li XF, Deng YQ, Tong YG, Qin CF. Excretion of infectious Zika virus in urine. Lancet Infect Dis. 2016;16(6):641–2.
Bonaldo MC, Ribeiro IP, Lima NS, Dos Santos AA, Menezes LS, da Cruz SO, de Mello IS, Furtado ND, de Moura EE, Damasceno L, et al. Isolation of infective Zika virus from urine and saliva of patients in Brazil. PLoS Negl Trop Dis. 2016;10(6):e0004816.
Atkinson B, Hearn P, Afrough B, Lumley S, Carter D, Aarons EJ, Simpson AJ, Brooks TJ, Hewson R. Detection of Zika virus in semen. Emerg Infect Dis. 2016;22(5):940.
Mansuy JM, Dutertre M, Mengelle C, Fourcade C, Marchou B, Delobel P, Izopet J, Martin-Blondel G. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect Dis. 2016;16(4):405.
Dupont-Rouzeyrol M, Biron A, O'Connor O, Huguon E, Descloux E. Infectious Zika viral particles in breastmilk. Lancet. 2016;387(10023):1051.
Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, Stanfield SM, Duffy MR. Genetic and serologic properties of Zika virus associated with an epidemic, yap state, Micronesia, 2007. Emerg Infect Dis. 2008;14(8):1232–9.
Acknowledgements
We are grateful to Dr. Robert Tesh (World Reference Center for Emerging Viruses and Arboviruses, The University of Texas Medical Branch, Galveston, USA) and Dr. Li-Sze Lim (Medical Innovation Ventures Pte. Ltd., Malaysia), for kindly providing us with Zika viruses.
Funding
This study was supported in parts by the Ministry of Education Malaysia under Long Term Research Grant Scheme (LRGS MRUN Phase 1: LRGS MRUN/F1/01/2018) and for niche area research under the Higher Institution Centre of Excellence (HICoE) Program (Project MO002–2019), the European Union’s Seventh Framework Programme (FP7/2007–2013) under the grant agreement 282589 – DengueTools, the Universiti Malaya (RU002–2019 and BK055–2017) and the Universiti Malaya Centre of Excellence (UMCoE) Top 100 Research Grant: UM.00000188/HGA.GV. The funders had no role in the design of the study, the collection, analysis and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
B.T.T and S.A.B. designed the study. B.T.T, K.L.C, N.I.S., S.K.L., S.S.S., K.K.T., C.S.K., J.A.J. and N.Z. conducted the experiments. B.T.T., S.S.S., K.K.T. and N.I.S. analysed the results. B.T.T., S.K.L., A.W.S., K.Z. and S.A.B. wrote the manuscript. The authors read and approved the final manuscript.
Authors’ information
SAB is a senior professor and director of the Tropical Infectious Diseases Research and Education Centre (TIDREC) at Universiti Malaya. He is also the director the WHO Collaborating Centre for Arbovirus Reference & Research at the Universiti Malaya. His research focus is on emerging infectious diseases particularly vector-borne and zoonotic diseases in the tropics. BTT is a senior lecturer in the TIDREC at Universiti Malaya. His research interest is on infectious diseases and laboratory diagnostics.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study obtained IRB ethics approval from the Universiti Malaya Medical Centre (UMMC) Medical Ethics Committee (Ethics Committee/IRB Reference Number: 908.11). Written consents have been obtained from all healthy donors who took part in the study.
Consent for publication
Not Applicable.
Competing interests
All authors declare no competing financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Preparation of the simulated clinical samples used in this study. A total of 24 simulated clinical samples consisting 8 saliva, 8 urine and 8 serum samples were prepared.
Additional file 2:
Figure S1. Map of RT-LAMP primers in alignment with the ZIKV NS2-NS3 gene junction consensus sequences. The nucleotide positions refer to the published complete genome of ZIKV MR766 (GenBank accession number: NC_012532).
Additional file 3:
Table S2. The ZIKV genomes used in this study.
Additional file 4:
Figure S2. Nucleotide mismatches of the RT-LAMP primers against 463 ZIKV genome sequences. The top sequences are primer sequences. Total numbers of ZIKV genomes with 100% sequence similarity to the primers are shown at 3′’ end of the primers. Only the ZIKV genomes with nucleotide mismatches to the primer are listed. The dots indicate the same nucleotides as the top sequence. The boxes indicate the nucleotide positions which are critical for priming and amplification.
Additional file 5:
Table S3. Time threshold of positivity for RT-LAMP assays of serially diluted ZIKV RNA.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Teoh, BT., Chin, KL., Samsudin, NI. et al. A reverse transcription loop-mediated isothermal amplification for broad coverage detection of Asian and African Zika virus lineages. BMC Infect Dis 20, 947 (2020). https://doi.org/10.1186/s12879-020-05585-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-020-05585-4