
Abstract
Background
Fungal bloodstream infections (BSI) may be serious and are associated with drastic rise in mortality and health care costs. Candida spp. are the predominant etiological agent of fungal sepsis. The prompt and species-level identification of Candida may influence patient outcome and survival. The aim of this study was to develop and evaluate the CanTub-simplex PCR assay coupled with Tm calling and subsequent high resolution melting (HRM) analysis to barcode seven clinically relevant Candida species.
Methods
Efficiency, coefficient of correlation and the limit of reliable detection were estimated on purified Candida EDTA-whole blood (WB) reference panels seeded with Candida albicans, Candida glabrata, Candida parapsilosis, Candida tropicalis, Candida krusei, Candida guilliermondii, Candida dubliniensis cells in a 6-log range. Discriminatory power was measured on EDTA-WB clinical panels on three different PCR platforms; LightCycler®96, LightCycler® Nano, LightCycler® 2.0. Inter- and intra assay consistencies were also calculated.
Results
The limit of reliable detection proved to be 0.2–2 genomic equivalent and the method was reliable on broad concentration ranges (106–10 CFU) providing distinctive melting peaks and characteristic HRM curves. The diagnostic accuracy of the discrimination proved to be the best on Roche LightCycler®2.0 platform. Repeatability was tested and proved to be % C.V.: 0.14 ± 0.06 on reference- and % C.V.: 0.14 ± 0.02 on clinical-plates accounting for a very high accuracy. Reproducibility was % C.V.: 0.11 between reference- and % C.V.: 0.12between clinical-panels which is highly acceptable.
Conclusion
Our assay demonstrates recent advances on Tm calling and HRM analysis for the molecular identification of relevant Candida species. This unique, simplex PCR assay may be capable to outperform conventional phenotypic methods by reducing time and providing accurate and reliable results directly from blood (2 h) or from whole blood culture bottles (12–24 h).
Similar content being viewed by others


Background
Fungal bloodstream infections may be serious and are associated with drastic rise in mortality and health care costs [1, 2]. Currently, with the gradually growing number of immunocompromised patients, we face an appreciable shift in the spectrum of fungal infections [3, 4]. The genus Candida corresponds to the most important cause of opportunistic mycoses in the world [5, 6]. Despite the antifungal therapy nosocomial Candida infections constitute a public health problem contributing to prolonged hospitalization time, generating enormous excess of costs for patient treatment, and a high mortality (25–60%) especially when complicated with septic shock [7, 8].
Candida spp. account for 70–80% of invasive fungal BSIs representing the third most frequent (all over 8–10%) cause of all BSIs in the intensive care unit (ICU) [9,10,11,12,13]. Nearly 95% of invasive candidemias are caused by Candida albicans, Candida glabrata, Candida parapsilosis, Candida tropicalis and Candida krusei [14,15,16]. The remaining 5% are caused by 10–12 species, such as C. guilliermondii, C. dubliniensis provoking superinfections due to their low sensitivity to broad-spectrum antifungals [10, 17]. The incidence of infections due to non-albicans species is continuously growing [13, 17]. Candida BSIs do not present with species specific clinical manifestations or laboratory abnormalities, thus the clinical course of the disease and the outcome are comparable in patients with sepsis caused by C. albicans and non-albicans species [12, 18].
Timely diagnosis of sepsis due to candidemia is essential for effective therapy, while delays of more than 12 h in the administration of antifungal drugs may substantially increase mortality [19,20,21,22,23].
The rapid and correct identification of Candida species can narrow therapy options by preventing treatment with potentially toxic antifungal agents, thus reducing costs of hospitalization and improving negative patient outcomes [24,25,26]. Because of the phenotypic similarities of Candida species, the turn-around time of traditional, culture based identification methods may take 2 to 8 days, delay adequate diagnosis and appropriate antifungal treatment. In comparison the surrogate-marker based molecular assays requiring an average of 3–4 h [27]. Nevertheless in some cases the morphological identification of clinically relevant Candida species is hampered by several difficulties, as is the case with the germ tube positive C. albicans and the potentially fluconazole-resistant, cryptic C. dubliniensis providing light green vs. dark green colonies on CHROM agar presenting only subtle differences in colony color [28, 29].
With regard to surrogate-marker based molecular methods, the antimannan antibody (Platelia GM-enzyme immunoassay, Bio-Rad) and 1,3-ß-D-glucan (BDG) antigen based (Fungitell assay, Associates of Cape Cod) immunological assays have good performances and may be useful in diagnosing of invasive fungal infections [30]. Nonetheless, as the panfungal BDG is a major cell wall component of Candida and other fungi, except Cryptococci and Zygomycetes, it not suitable for the identification even to the genus level [12].
Along with the growing number of annotated protein spectra and the development of refined and standardized methodologies of protein extraction methods, matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) has emerged as a powerful technology for the prompt identification of microorganisms in clinical microbiology supporting genus-, or species-oriented treatment [31,32,33].
The DNA sequencing methods, targeting the 18S rRNA genes or ITS regions, can generate accurate species-level identification for many microbial isolates; however these methods are time consuming and technically demanding.
Polymerase chain reaction (PCR)-based techniques appear to be promising in terms of speed, economy, and resolution power [34]. PCR based patient follow up were shown to be capable to precede clinical signs of invasive candidiasis (IC) with the range of 1 day to 4 weeks, furthermore treatment could be initiated 3 days (range: 0–8 days) before the blood culture diagnosis was taken [35]. There have been numerous platforms and gene targets used for Candida spp. identification, such as those genes encoding cytochrome P450, actin and L1A1, and the highly variable internal transcribed spacer regions; ITS1 and ITS2 in combination with the relatively conserved regions of 18S, 5.8S or 28S nuclear rRNA genes [36,37,38,39,40,41].
MALDI-TOF and real-time PCR applications have high throughput, low-cost in supplies, short turnaround time. Both tools have the advantage to identify a broad-range of clinically relevant pathogenes with high accuracy at the species level Though MALDI-TOF is not suitable to detect a low amount of microorganisms directly from blood and PCR techniques usually lack standardization.
The prime aim of this study is to develop and evaluate a real-time PCR method which can identify and differentiate among seven relevant Candida spp. with high accuracy in a single, closed tube system, by a post-PCR Tm calling assay coupled with a contingent high-resolution melting (HRM) analysis. This barcoding method is a single locus based HRM system which relies on the conserved regions of Candida beta-tubulin genes permitting reliable and precise species-specific identification of seven reference strains and 38 clinical isolates. To our knowledge, there is no other published method which is tailored to a single domain of Candida beta-tubulin genes and there is no other conventionally available or presented simplex PCR assay.
Methods
Fungal and bacterial strains
Genomic DNA (gDNA) samples of 81 clinically relevant fungal strains; Candida (43), aspergilli (32), Fusarium (4), Lichtemia (1), Rhyzopus (1), Scedosporium (1) and 16 bacteria; Gram-positive (10) and Gram-negative (6) were examined. The reference strains and clinical isolates (Table 1) were maintained at the Department of Microbiology, University of Szeged and at the Department of Medical Microbiology, University of Debrecen. To preserve the viability and purity of fungal and bacterial strains they were maintained in 50% glycerol stock solution at − 80 °C and were periodically subcultured.
DNA extraction
All genomic DNA (gDNA) extraction steps were performed in a class II laminar-flow cabinet to avoid environmental contamination.
Fungal gDNA extraction
Candida cultures used for the molecular identification of the strains were grown on yeast-peptone D-glucose (YPD) for 2 days, and DNA was extracted using the Masterpure™ Yeast DNA Purification Kit (Epicentre Biotechnol., Madison, USA) per the manufacturer’s instructions [42]. Mould reference strains (Aspergillus only) and clinical isolates (Aspergillus, Fusarium, Lichtemia, Rhyzopus, Scedosporium) were cultivated on standard minimal nitrate medium [43]. DNA extractions were carried out at the University of Debrecen (Department of Biotechnology and Microbiology). gDNA was isolated from liquid cultures grown in minimal medium at 37 °C (A. fumigatus, A. niger), 25 °C (A. terreus, A. lentulus, A. flavus) and 30 °C (A. welwitschiae) at 220 rpm for 18 h. The mycelium was disrupted by using Roche MagNa Lyser (Roche Diagnostics, Risch-Rotkreuz, Switzerland) and gDNA was isolated using the Genomic DNA Purification Kit (Thermo Scientific, Maryland, USA) per the manufacturer’s instructions.
Bacterial gDNA extraction
The bacterial strains were grown on Müller-Hinton agar base under aerobic conditions. DNA was extracted using the E.Z.N.A.® Bacterial DNA Kit (Omega Biotech, Norcross, Georgia, USA) per the manufacturer’s instructions. Briefly, for suspension cultures one millilitre of log phase culture with approximately 108 bacterial cells was used and cells were pelleted by centrifugation. Gram-positive bacterial cell walls were lysed by lysozyme solution with 20 mg ml− 1 enzyme in 20 mM Tris HCL, pH 8.0 2 mM EDTA, 1.2% TritonX100).
DNA concentrations and purity were measured using NanoDrop-1000 spectrophotometer (NanoDrop Technologies, Inc., North Carolina, USA).
CanTub-simplex PCR
Annotated sequences of the Candida beta-tubulin genes were extracted from the EMBL/GeneBank databases to make multiple alignments using Clustal Omega. Available Candida beta-tubulin sequences were aligned surveying potential sequence deviations within species. When designing primers, we screened for melting domains covering enough mismatches to enable proper discrimination among the tested strains but comprising 30–40 nucleotide long conserved nucleotides on flanking regions for the very specific hybridization of primers and avoiding cross-reactions with gDNA of other BSI causing fungal, bacterial strains and with the human genomic DNA.
The CanTub forward primer is 37 bp long(5’-CTAAAATCAGAGAAGAATTCCCCTGATAGAATGATGGC-3′), GC content 37.8%, Tm 73.2 °C and the CanTub reverse primer is 43 bp long (5’-CAATTGACCTGGGATAACGTAAAGAAGTAGTAACACCAGACAT-3′), GC content 38%, Tm 74.2 °C.
Setting the optimal CanTub-simplex PCR conditions
Temperature gradient assay was performed from 55 to 72 °C for assessing the performance of the primer pair during amplification with a temperature gradient program using the LightCycler® 96 thermal cycler Instrument (Roche Applied Science, Penzberg, Germany). The MgCl2 optimization was performed by adding different amounts of MgCl2 within the concentration range 1 to 3.5 mM. The primer optimization assay was performed using 0.2, 0.4, 0.6 μM of the forward and reverse primers.
Verification of the CanTub-simplex amplicons via conventional PCR
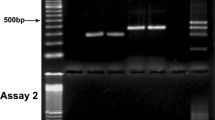
In order to monitor the accumulation of aspecific amplicons in the CanTub-simplex PCR, primers were tested with 20 ng gDNA of five Candida reference strains (C. albicans ATCC 10231, C. glabrata ATCC 90030, C. parapsilosis ATCC 22019, C. tropicalis ATCC 750, C. krusei ATCC 6258), and two clinical isolates (C. guilliermondii SZMC 1536 and C. dubliniensis SZMC 1470 depicted by ID33 and ID36 in Table 1). The thermocycling reactions were conducted in a Roche LightCycler®Nano instrument (P2). 200 ng of the yielded CanTub PCR amplicons were electrophoresed on 2% TBE-agarose gel stained with ethidium-bromide to investigate aspecific PCR increments.
Real time PCR platforms
Real-time PCR assays were conducted on three different PCR platforms (P1, P2, P3). (P1) LightCycler® 96 thermal cycler, (P2) LightCycler® Nano and (P3) LightCycler® 2.0 Instruments (Roche Diagnostics, Risch-Rotkreuz, Switzerland) were used with the LightCycler 480 High-Resolution Melting Master (Roche Applied Science, Penzberg, Germany) (P1) 10 μl reaction volumes consisted of 5 μl 2× LightCycler 480 High Resolution Melting Master, 0.5 μl of each primer (0.4 μM), 1 μl MgCl2 (2 mM) and 3 μl template DNA. The thermocycling reactions were conducted in a LightCycler 480 Multiwell Plate, (white). (P2), (P3) 20 μl reaction volumes consisted of 10 μl 2× LightCycler 480 High Resolution Melting Master, 0.5 μl of each primer (0.4 μM), 5 μl PCR grade water, 1 μl MgCl2 (2 mM) and 3 μl template DNA. The thermocycling reactions were conducted in LightCycler® 8-Tube Strips (P2) and in 20 μl glass capillaries (P3). The real-time PCR runs always included at least two controls of reaction mix without DNA (non-template control - NTC). Temperature parameters were set as follows: an initial denaturing step of 95 °C for 10 min followed by 50 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 10 s and extension at 72 °C for 10 s. Fluorescent data were collected in the ResolightDye channel (470/514 nm).
Melting temperature calculation and HRM protocols
The accumulation of the Candida species descriptive amplicons was monitored via melting temperature calculation and HRM analysis on three real-time PCR platforms.
(P1), (P2) Following the completion of real-time PCR, the products were denatured at 95 °C for 15 s (4.4 °C s− 1), and then renatured at 40 °C for 15 s (1 °C s− 1) to form DNA duplexes. HRM analysis was performed by increasing the temperatures from 45 to 97 °C (0.2 °C s− 1) recording changes in fluorescence (-dF/dT) and plotting against changes in temperature. The HRM profiles were then analyzed using the LightCycler®96 HRM analysis and the LightCycler® Nano Software thus highly dense measurement points were taken during the high resolution melting stage resulting in species descriptive HRM melting curves.
(P3) HRM analysis was not performed with the LC 2.0 software due to the lack of this option. For Tm analysis melting temperature measurement was set from 45 to 97 °C (0.05 °C s− 1). Melting peaks were analyzed by the LightCycler®2.0 Software.
Cross reactivity
Possible cross reactions of the CanTub-HRM assay were tested with approximately 20–25 ng purified (OD260/2801.82–1.97) gDNA of human placenta (Sigma Aldrich, Missouri, USA) and the gDNA samples of 32 aspergilli; 6 reference strains (Aspergillus terreus FGSC A1156, A. fumigatus Af293, A. lentulus CBS117885, A. flavus NRRL11611, A. niger CBS 113.46, A. tubingensis CBS 134.48), further 26 gDNA of Aspergillus (ID39–64), Fusarium (ID65–68), Lichtemia corymbifera (ID69), Rhyzopus oryzae (ID70), Scedosporium aurantiacum (ID71) clinical isolates, finally with the gDNA of 10 Gram-positive bacteria; 5 reference strains (Staphylococcus aureus ATCC 25923, 29,213, 43,300, Enterococcus faecalis ATCC 29212, Enterococcus hirae ATCC 8043), 5 Gram-positive clinical isolates (ID72–76) and with 6 Gram-negative bacteria; Enterobacter aerogenes ATCC13048 reference strain and five Gram-negative clinical isolates (ID77–81) in Table 1.
Verification of the CanTub-simplex assay on EDTA-whole blood panels
Obtaining of EDTA-WB samples for spike in controls
Working with WB samples from healthy volunteers was approved by the local ethics committee, MK-JA/50/0096–01/2017. Participants agreed to take part in this study. EDTA whole blood samples obtained from healthy volunteers were pooled and screened for Candida contamination prior to use as extraction negative controls (ENC).
Candida EDTA-WB reference panels
To obtain Candida reference panels in a 6-log range (Candida albicans ATCC 10231; ref-panel_1, C. glabrata ATCC 90030; ref-panel_2, C. parapsilosis ATCC 22019; ref-panel_3, C. tropicalis ATCC 750; ref-panel_4, C. krusei ATCC 6258; ref-panel_5, C. guilliermondii SZMC1536; ref-panel_6, C. dubliniensis SZMC 1470; ref-panel_7), 990 μl of EDTA-WB samples were seeded with 10 μl of log-phase culture suspensions at a cell density of 108–102 CFU in one millilitre of yeast suspensions. Following DNA purification the sample extracts contained 2 × 105 (106 CFU/1 ml WB) – 0.2 (10 CFU/1 ml WB) GE in 3 μl of PCR template.
Candida EDTA-WB clinical panels
EDTA-WB samples at a cell density of 104 CFU/ml Candida strains (ID1–38) were used to perform seven Candida clinical panels (ID1–7; clin-panel_1, ID8–11; clin-panel_2, ID12–17; clin-panel_3, ID18–24; clin-panel_4, ID25–30; clin-panel_5, ID31–34; clin-panel_6, ID35–38; clin-panel_7). Following DNA purification the sample extracts contained 200 GE in 3 μl of PCR template.
DNA purification from EDTA-WB samples
DNA purification steps were performed in a class II laminar air-flow cabinet to avoid environmental contamination. Spiked EDTA-WB samples were disrupted (2000 rpm, 1 min) by Roche MagNa Lyser (Roche Diagnostics, Risch-Rotkreuz, Switzerland), and DNA was extracted along with the ENCs using the High Pure Viral Nucleic Acid Large Volume Kit (Roche Applied Science) according to manufacturer’s instructions. To obtain technical duplicates every sample extractions were performed in parallel. The elution volumes were adjusted to 15 μl and pooled.
PCR efficiency and the limit of reliable detection (LoRD)
Amplification reactions were carried out on three different PCR platforms; P1, P2, P3. Duplicate PCRs were performed at every dilution of the Candida EDTA-WB reference panel extracts. Quantification cycle (Cq) values were subtracted and melting curves were analyzed to estimate the lowest template DNA concentration by which the appropriate, species descriptive melting peaks were interpretable (LoRD). To study the correlation between the Cq-s and the genomic load standard curves were obtained by plotting Cq values against the log of cell number; 106–10 CFU/1 ml WB which is equivalent to 2 × 105–0.2 GE/PCR. Mean Cq data were calculated and standard curves were built where these plots determined the linear dynamic ranges. Efficiency was calculated per the following formula, E = (10–1/slope) than was converted to percentage efficiency by using the formula, E% = (E-1) × 100 [44]. Amplification efficiency, (correlation coefficient; R2) was also calculated.
In-house quality assessment
PCRs were conducted on P1 and Tm data were subtracted.
Determining the repeatability
To estimate repeatability intra-assay consistency was calculated. To calculate the coefficient of variation (% C.V.) of the CanTub-simplex PCR within the three PCR plates (ref-plate_1, _2, _3) of the seven Candida reference panel extracts (ref-panel_1-_7) and within the four PCR plates (clin-plate_1, _2, _3, _4) of the seven Candida clinical panel extracts (clin-panel_1-_7) standard deviation (±SD) of duplicate Tm mean data were taken, dividing these numbers by the mean of the Tm values and multiplying them by 100 [see Additional file 1]. Finally, the grand mean of the sample coefficient of variations (average % C.V.-s) of the three plates was taken defining the plate % C.V. values. When the intra-assay % C.V. is below 10% the method under investigation has high precision.
Determining the reproducibility
To estimate the precision of the CanTub-simplex PCR as regards discrimination of the relevant Candida species and to confirm that results generated are consistent over time inter-assay consistency (plate-to-plate variation) was estimated between the PCR plates on three distinct days with the seven Candida reference panel extracts and with the sample extracts of the seven Candida clinical panels [see Additional file 2]. In the case of the Candida panels duplicate PCRs were performed on every sample isolate. Tm data were subtracted than mean Tm (°C) with standard deviations (±SD) were calculated. When analyzing Candida clinical panels, duplicate Tm means of adherent clinical strains of the different species were assembled and overall mean was calculated. Plate coefficient of variations (% C.V.) was calculated. Finally, grand mean of the sample coefficient of variations (average % C.V.-s) was taken. Inter-assay % C.V. values less than 15% are generally acceptable.
Results
In silico assessment of the CanTub forward and CanTub reverse primers
Based on the sequence alignment data we presumed that the amplicons generated by CanTub-simplex primers will display enough sequence divergence between closely related Candida species but at the same time may be conserved enough barcoding the different clinical isolates within species. Primer matches with annotated beta-tubulin genes from C. albicans, C. tropicalis, C. dubliniensis and C. glabrata are indicated in [see Additional file 3] with the sizes of the amplicons.
Optimal reaction conditions with the CanTub-simplex PCR
Optimal reaction conditions were determined as described in the methods section. 0.4 μM primers-, 2 mM MgCl2-concentrations and annealing at 60 °C proved to be optimal. The size of the amplified CanTub-simplex PCR bands were verified on five Candida reference strains and on two clinical isolates. All the amplified CanTub-simplex PCR fragments are smaller than 300 bp in size and we did not experience any aspecific PCR increments (data not shown).
Cross reactivity
No cross-amplification was detected with the human genomic DNA and with the Gram-negative and Gram-positive bacteria. Furthermore, no cross-amplification was observed with A. terreus, A. lentulus, A. tubingensis, A. viridinutans and A. udagawae strains. Mild cross reaction was detected with the A. fumigatus, A. flavus and A. niger providing Tm peaks at 86.35 ± 0.16 °C and displaying inconclusive HRM curves and peaks. Moderate false positivity with plain, inconsistent peaks were detected in the case of Rhyzopus oryzae (77.38 °C), Scedosporium aurantiacum (75.73 °C) generating cycle threshold (Cq) values greater than Cq > 42 (data not shown).
Analytical sensitivity
LoRD of the simplex CanTub-PCR assay was estimated on Candida EDTA-WB standard panel extracts containing serially diluted Candida gDNA in a 6-log range. Linear dynamic ranges were assessed on P1, P2, and P3 with PCR efficiency (E) and coefficient of correlation (R2) were also estimated using the standard curve data (Fig. 1). The analytical sensitivity with the lowest concentration of template DNA where reliable identifications with conclusive melting peaks and HRM curves were attainable in the case of at least one technical duplicate proved to be 0.2 GE/PCR (10 cells/1 ml WB) on all panels and on all platforms except the C. parapsilosis reference panel on P1 and P3, where LoRD proved to be 2 GE/PCR (100 cells/1 ml WB) only. On P2 the LoRD was 2 GE/PCR also for C. parapsilosis, C. dubliniensis and C. guilliermondii reference panels.

Regression lines of CanTub-simplex PCR on P1-P3 platforms. The limit of reliable detection (LoRD) was evaluated on seven, purified Candida EDTA-WB reference panels of the Candida albicans (ATCC 10231), Candida glabrata (ATCC 90030), Candida parapsilosis (ATCC 22019), Candida tropicalis (ATCC 750), Candida krusei (ATCC 6258) type strains and the Candida guilliermondii (SZMC 1536) and Candida dubliniensis (SZMC 1470) clinical isolates. Y-intercept, slope, PCR reaction efficiency (E%), error (±), coefficient of correlation (R2), and limit of reliable detection (LoD) measured in 20 μl PCR reactions are shown
Maximum discriminatory property was achieved by defining seven melting clusters on three real-time platforms
CanTub-simplex assay targeting the beta-tubulin genes proved to be accurate and reliable for specific identification of seven Candida species. Thirty eight clinical isolates phenotypically identified as Candida species were subsequently tested in a blinded manner and barcoded due to comparing the melting data to those of the reference strains.
Normalized melting curves (Fig. 2/a) with the difference plot (Fig. 2/b) are shown by using saturating concentrations of a fluorescent double stranded DNA intercalating ResoLight® dye. HRM analyses were performed on Candida EDTA-WB clinical panels and on platform P1.

Discriminatory property of the CanTub-simplex PCR. Species level identification was achieved by using CanTub-simplex PCR primer pair to accurately identify Candida species based on HRM analysis (P1) by using the LightCycler®96 HRM analysis Software (Roche Diagnostics, Risch-Rotkreuz, Switzerland). a Representation of the normalized CanTub-simplex PCR melting curves. Each colour represents a different species: Candida albicans (blue) ID1–7; Candida glabrata (red) ID8–11; Candida parapsilosis (yellow) ID12–17; Candida tropicalis (orange) ID18–24; Candida krusei (green) ID25–30; Candida guilliermondii (brown) ID31–34; Candida dubliniensis (pink) ID35–38. b Representation of the normalized difference plots. Normalized difference plots against Candida albicans
Whisker plots show the distribution of the Tm values when testing the 38 Candida clinical strains on four separate plates. For every dataset, the median, minimum, maximum Tm values along with the 25th and 75th percentile are also shown in Fig. 3. Based on the distribution of the melting temperature data of the Candida strains tested, we created seven melting clusters for the reliable species level identification on P1-P3 C. dubliniensis cluster_1 Tm with 95% CI: 77.58 ± 0.32 °C (P1), 78.39 ± 0.22 °C (P2), 76.45 ± 0.37 °C (P3). C. tropicalis cluster_2 Tm with 95% CI: 77.96 ± 0.2 °C (P1), 78.77 ± 0.21 °C (P2), 77.09 ± 0.41 °C (P3). C. albicans cluster_3 Tm with 95% CI: 78.64 ± 0.18 °C (P1), 79.16 ± 0.14 °C (P2), 77.61 ± 0.15 °C (P3). C. krusei cluster_4 Tm with 95% CI: 79.17 ± 0.26 °C (P1), 78.64 ± 0.18 °C (P2), 78.17 ± 0.24 °C (P3). C. parapsilosis cluster_5 Tm with 95% CI: with 95% CI: 79.92 ± 0.37 °C (P1), 80.08 ± 0.3 °C (P2), 78.65 ± 0.39 °C (P3). C. guilliermondii cluster_6 Tm with 95% CI: 80.98 ± 0.3 °C (P1), 81.19 ± 0.3 °C (P2), 79.76 ± 0.18 °C (P3). C. glabrata cluster_7 Tm with 95% CI: 81.3 ± 0.31 °C (P1), 81.6 ± 0.32 °C (P2), 82.02 ± 0.32 °C (P3).Using these melting clusters for PCR typifying of unknown Candida strains there is no need for using standards as reference controls.

Whisker plots of the Candida species descriptive clusters. Whisker plots representing the discriminatory power of the CanTub-simplex PCR were created based on the digitalized melting temperature (Tm) data representing the seven melting clusters (pink cluster_1 for Candida dubliniensis, orange cluster_2 for Candida tropicalis, blue cluster_3 for Candida albicans, green cluster_4 for Candida krusei, yellow cluster_5 for Candida parapsilosis, brown cluster_6 for Candida guilliermondii, red cluster_7 for Candida glabrata) for the discrimination of the Candida species. Melting peaks were subtracted from the analysis of purified the Candida EDTA-WB clinical panel extracts (P1-P3). Tm data of adherent clinical strains of the different species were assembled and data were grouped. Whisker plots were constructed for each data group showing the range of obtained temperatures of melting (Tm); the minimum, the median, and the maximum Tm values with 25th and 75th percentiles
Repeatability and reproducibility
We estimated the intra-, and inter-assay consistency of the CanTub-simplex PCR on Candida EDTA-WB reference- [see Additional file 1] and on Candida EDTA-WB clinical panels [see Additional file 2]. For measuring the precision of the assay average coefficient of variation was calculated for the technical duplicate Tm values measured on P1 where intra-assay % C. V. proved to be 0.14 (ref. plate_1), 0.19 (ref. plate_2) and 0.08 (ref. plate_3) on Candida EDTA-WB reference plates and 0.14 (clin. plate_1), 0.12 (clin. plate_2), 0.10 (clin. plate_3), 0.13 (clin. plate_4) on Candida EDTA-WB clinical plates accounting for a very high accuracy of the CanTub-simplex PCR. Plate-to-plate consistency was also assessed for the assay between the three Candida EDTA-WB reference plates and the four Candida EDTA-WB clinical panels. Reproducibility was measured and sample coefficient of variations (grand % C.V.-s) between the three reference and four clinical plates were taken, where inter-assay proved to be 0.11between standard [see Additional file 1] and 0.12between clinical panels [see Additional file 2] which is highly acceptable.
Discussion
With the continuously growing population of immunocompromised patients, the number of invasive fungal infections has increased significantly over the past decades [1,2,3,4]. Among all, Candida species are the most prevalent etiology of sepsis and septic shock in critically ill patient groups representing a significant health challenge with increasing medical and economic importance [7,8,9]. Concern is rising about the growing incidence of non-albicans infections and the emergence of their various intrinsic or acquired resistances. Over the last two decades, the occurrence of non-albicans species have emerged [12, 13, 17, 18, 26]. More than 98% of candidemia cases are caused by C. albicans, C. glabrata, C. parapsilosis, C. tropicalis, C. krusei, including further species such as C. dubliniensis, and C. guillermondii [14,15,16].
Identification of Candida species is important due to the differences in the antifungal susceptibility profile associated with species and because of the limitation of the phenotypic identification [10,11,12,13,14,15,16,17].
The traditional culture based morphological (colony and microscopic morphology), and/or biochemical (such as sugar assimilation and fermentation tests) identification methods of the Candida species are laborious, requiring a high level of skills and expertise of clinical mycologists and a long period of time, limiting patient care [27, 28]. Morphological identification of certain species remains problematic due to the high degree of phenotypic similarities between C. albicans versus C. tropicalis and C. dubliniensis out of which the latter species can acquire stable fluconazole resistance rapidly [28].
There is an increasing demand for innovative sensitive, rapid and non-invasive methods for identification of Candida species at an early stage of the disease.
Tm calling assays especially when coupled with HRM analysis has been introduced for scanning genotypes and for the rapid discrimination between DNA sequences based on their variant high resolution melting profiles without the use of fluorescent labeled probes. Although HRM based methods do not have the resolving power that many sequencing techniques have and are not as sensitive as the TaqMan probe based systems, they became more and more attractive to molecular diagnostic laboratories. Our recent data also support that applications resting on HRM analyses may be ideally suited for identifying or ruling out certain fungal and bacterial pathogens in clinical diagnostic workflow.
In 2007 Carvalho and colleagues developed and tested on 231 Candida isolates a sensitive (2.15 ± 0.25 CFU/1 ml), multiplex PCR based method by using yeast specific (ITS1, ITS2) universal and species-specific (18S, 28S) primers allowing the barcoding of eight relevant Candida strains (C. albicans, C. glabrata, C. parapsilosis, C. tropicalis, C. krusei, C. guilliermondii, C. lusitaniae and C. dubliniensis) at the species level taking the advantage of the presence of high-copy number of rRNA genes due to the presence of characteristic PCR amplified band patterns [36]. The only drawback of this method is that barcoding of unknown yeasts require running eight simultaneous PCR assays.
The CanTub-simplex PCR assay described here relies on a single primer pair targeting specific regions of Candida beta-tubulin genes.
The assay provides reliable nucleic acid based testing for proper identification of seven relevant Candida species by defining the species-specific melting domains and/or the shape of the derivative melting curves on the three real-time platforms (P1-P3).
When analyzing 1 ml of WB samples infected with 106–10 Candida cells the amplification efficiency was 100% on all reference panels (ref. panel_1 – ref. panel_7) observing efficient amplification and melt curve analysis. For samples seeded with 1 CFU sample the barcoding capacity proved to be only 78.57%. Nevertheless, on all Candida reference panels the amplification efficiencies were reliable (E% = 1.85–1.94). It must be also noted, that the standard deviations (mean SD: ±0.12 °C) of the Tm data measured on reference panels proved to be low even in bright concentration ranges.
Here we demonstrated that when appropriate DNA samples are available (OD260/2801.82–1.97 and gDNA concentration of 1–10 ng/μl), CanTub-simplex PCR assay identifies the seven most frequent pathogenic Candida species. CanTub-simplex PCR based applications used in parallel with morphotyping may offer better resolution of the species level identification.
This method is technically not demanding and provides clear, easily traceable protocols on three different real-time platforms. Single locus primer pair is used so the method can be easily multiplexed with exogenous or endogenous TaqMan internal control assays. Here we also demonstrated that the adaptation of DNA based applications used in tandem with morphological examinations in clinical diagnostic laboratories can offer better resolution of species within the genus.
Conclusion
CanTub-simplex PCR targeting the beta-tubulin genes proved to be sensitive and accurate on all platforms (P1 – P3) tested here, and has the potential to identify seven clinically relevant Candida species (C. albicans, C. glabrata, C. parapsilosis, C. tropicalis, C. krusei, C. guilliermondii, C. dubliniensis) by using the seven, pre-defined melting clusters without using reference controls. These features make this CanTub-simplex PCR advantageous for use as a first-pass diagnostic adjunct in microbiology laboratories on blood culture bottles or for direct whole blood testing by making the patient follow-up more achievable.
Abbreviations
- % C.V:
-
Coefficient of variation
- ±SD:
-
Standard deviation
- ATCC:
-
American type culture collection
- BDG:
-
1,3-ß-D-glucan
- bp:
-
Base pairs
- BSI:
-
Bloodstream infections
- CanTub:
-
Candida tubulin
- CBS:
-
Centraalbureau voor Schimmelcultures
- CFU:
-
Colony forming unit
- Cq:
-
Quantification cycle
- E:
-
Efficiency
- EDTA:
-
Ethylenediaminetetraacetic acid
- ENC:
-
Extraction negative controls
- gDNA:
-
Genomic DNA
- GE:
-
Genomic equivalent
- HRM:
-
High resolution melting
- IC:
-
Invasive candidiasis
- ICU:
-
Intensive care unit
- ITS:
-
Internal transcribed spacer
- LoRD:
-
Limit of reliable detection
- MALDI-TOF:
-
Matrix assisted laser desorption/ionization time-of-flight
- MS:
-
Mass spectrometry
- NRRL:
-
Agricultural research service culture collection
- NTC:
-
Non-template control
- OD:
-
Optical density
- PCR:
-
Polymerase chain reaction
- rDNA:
-
Ribosomal DNA
- SZMC:
-
Szeged microbiology collection
- TBE:
-
Tris/Borate/EDTA
- Tm:
-
Melting temperature
- WB:
-
Whole blood
- YPD:
-
Yeast-pepton-D-glucose
References
Ramana KV, Kandi S, Bharatkumar VP, et al. Invasive fungal infections: a comprehensive review. Am J Infect Dis. 2013;4:64–9.
Shawn RL. Current epidemiology of Candida infection. J of Clin Microbiol Newsletter. 2014;36:131–6.
Falagas ME, Roussos N, Vardakas KZ. Relative frequency of albicans and the various non-albicans Candida spp. among candidemia isolates from patients in various parts of the world: a systematic review. Int J Infect Dis. 2010;14:e954–66.
Pfaller MA, Diekema DJ, Gibbs DL, et al. Results of the ARTEMIS DISK global antifungal surveillance study 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida species to fluconazol and voriconazole as determined by CLSI standardized disk diffusion. J. Clin. Microbiol. 2010;48:1366–77.
Cassia Orlandi Sardi J, de Souza Pitangui N, Gullo F, Soares Mendes Giannini MJ. A mini review of Candida species in hospital infection: epidemiology, virulence factor and drugs resistance and prophylaxis. Trop Med Surg. 2013;1:141.
Das I, Nightingale P, Patel M, Jumaa P. Epidemiology, clinical characteristics, and outcome of candidemia: experience in a tertiary referral center in the UK. Int J Infect Dis. 2011;15:e759–63.
Pappas PG, Rex JH, Lee J, et al. A prospective observational study of candidemia: epidemiology, therapy and influence on mortality in hospitalized adult and pediatric patients. Clin Infect Dis. 2003;37:634–43.
Hassan I, Powell G, Sidhu M, Hart WM, Denning DW. Excess mortality, length of stay and cost attributable to candidemia. J Inf Secur. 2009;59(5):360–5.
Yapar N. Epidemiology and risk factors for invasive candidiasis. Ther Clin Risk Manag. 2014;10:95–105.
Richardson M, Lass-Flörl C. Changing epidemiology of systemic fungal infections. Clin. Microbiol. Infect. 2008;Suppl 4:5–24.
Walsh TJ, Groll A, Heimenz J, Fleming R, Roilides E, Anaissie E. Infections due to emerging and uncommon medically important fungal pathogens. Clin. Microbiol. Infect. 2004;Suppl 1:48–66.
Delaloye J, Calandra T. Invasive candidiasis as a cause of sepsis in the critically ill patient. Virulence. 2014;5:161–9.
Selvarangan R, Bui U, Limaye AP, Cookson BT. Rapid identification of commonly encountered Candida species directly from blood culture bottles. J Clin Microbiol. 2003;41:5660–4.
Trick WE, Fridkin SK, Edwards JR, Hajjeh RA, Gaynes RP, National Nosocomial Infections Surveillance System Hospital. Secular trend of hospital acquired candidemia among intensive care unit patients in the United States during 1989-1999. Clin Infect Dis. 2002;35:627–30.
Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–17.
Hajjeh RA, Sofair AN, Harrison LH, et al. Incidence of bloodstream infections due to Candida species and in vitro susceptibilities of isolates collected from 1998 to 2000 in a population based active surveillance program. J Clin Microbiol. 2004;42:1519–27.
Sardi JC, Scorzoni L, Bernardi T, Fusco-Almeida AM, Mendes Gianni MJ. Candia species: current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J Med Microbiol. 2013;62:10–24.
Wisplinghoff H, Seifert H, Wenzel RP, Edmond MB. Inflammatory response and clinical course of adult patients with nosocomial bloodstream infecions caused by Candida spp. Clin Microbiol Infect. 2006;12:170–7.
Ortega M, Marco F, Soriano A, et al. Candida species bloodstream infection epidemiology and outcome in a single institution from 1991 to 2008. J Hosp Infect. 2011;77:157–61.
Horn DL, Neofytos D, Anaissie EJ, et al. Epidemiology and outcomes of candidemia in 2019 patients data from the prospective antifungal therapy alliance registry. Clin Infect Dis. 2009;48:1695–703.
Xafranski H, Melo AS, Machado AM, Briones MR, Colombo AL. A quick and low-cost PCR-based assay for Candida spp identification in positive blood culture bottles. BMC Infect Dis. 2013;13:467.
Garey KW, Rege M, Pai MP, et al. Time to initiation of fluconazole therapy impacts mortality in patients with candidaemia: a multi-institutional study. Clin Infect Dis. 2006;43:25–31.
Morrell M, Fraser VJ, Kollef MH. Delaying the empiric treatment of Candida bloodstream infection until positive blood culture results are obtained: a potential risk factor for hospital mortality. Antimicrob Agents Chemother. 2005;49:3640–5.
Pappas PG, Rex JH, Sobel JD, et al. Guidelines for treatment of candidiasis. Clin Infect Dis. 2004;38:161–89.
Slavin MA, Sorrell TC, Marriott D, et al. Candidemia in adult cancer patients: risks for fluconazole-resistant isolates and death. J Antimicrob Chemother. 2010;65:1042–51.
Pappas PG, Kauffman CA, Andes DR, et al. Clinical practice guideline for the management of candidiasis: 2016 update by infectious diseases society of America. Clin Infect Dis. 2016;62:e1–50.
Pfeiffer CD, Samsa GP, Schell WA, Reller LB, Perfect JR, Alexander BD. Quantitation of Candida CFU in initial positive blood cultures. J Clin Microbiol. 2011;49:2879–83.
Selvarangan R, Limaye AP, Cookson BT. Rapid identification and differentiation of Candida albicans and Candida dubliniensis by capillary-based amplification and fluorescent probe hybridization. J Clin Microbiol. 2002;40:4308–12.
Lockhart SR, Iqbal N, Cleveland AA, et al. Species identification and antifungal susceptibility testing of Candida bloodstream isolates from population-based surveillance studies in two U. S. cities from 2008–2011. J. Clin. Microbiol. 2012;50:3435–42.
Clancy CJ, Nguyen MH. Finding the “missing 50%” of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care. Clin Infect Dis. 2013;56:1284–92.
Pulcrano G, Lula DV, Vollaro A, et al. Rapid and reliable MALDI-TOF mass spectrometry identification of Candida non-albicans isolates from bloodstream infections. J Microbiol Methods. 2013;94:262–6.
Putignani L, Del Chierico F, Onori M, et al. MALDI-TOF mass spectrometry proteomic phenotyping of clinically relevant fungi. Mol BioSyst. 2011;7:620–9.
Saghrouni F, Abdeljelil JB, Boukadida J, Ben Said M. Molecular methods for strain typing of Candida albicans: a review. J Appl Microbiol. 2013;114(6):1559–74.
Trost A, Graf B, Eucker J, Sezer O, Possinger K, Göbel UB, Adam T. Identification of clinically relevant yeasts by PCR/RFLP. J Microbiol Methods. 2004;56(2):201–11.
Lin M-T, Lu H-C, Chen W-L. Improving efficacy of antifungal therapy by polymerase chain reaction-based strategy among febrile patients with neutropenia and cancer. Clin Infect Dis. 2001;33:1621–7.
Carvalho A, Costa-De-Oliveira S, Martins ML, Pina-Vaz C, Rodrigues AG, Ludovico P, Rodrigues F. Multiplex PCR identification of eight clinically relevant Candida species. Med Mycol. 2007;45(7):619–27.
Avni T, Leibovici L, Paul M. PCR diagnosis of invasive candidiasis: systematic review and meta-analysis. J Clin Microbiol. 2011;49:665–70.
Hsiue HC, Huang YT, Kuo YL, Liao CH, Chang TC, Hsueh PR. Rapid identification of fungal pathogens in positive blood cultures using oligonucleotide array hybridization. Clin Microbiol Infect. 2010;16:493–500.
Fidler G, Kocsube S, Leiter E, Biro S, Paholcsek M. DNA barcoding coupled with high resolution melting analysis enables rapid and accurate distinction of Aspergillus species. Med Mycol. 2017;55(6):642–59.
Balajee SA, Houbraken J, Verweij PE, et al. Aspergillus species identification in the clinical setting. Stud Mycol. 2007;59:39–46.
Nemcova E, Cernochova M, Ruzicka F, Malisova B, Freiberger T, Nemec P. Rapid identification of medically important Candida isolates using high resolution melting analysis. PLoS One. 2015;10:e0116940.
Barratt RW, Johnson GB, Ogata WN. Wild-type and mutant stocks of Aspergillus nidulans. Genetics. 1965;52:233–46.
Liu D, Coloe S, Baird R, Pedersen J. Rapid mini-preparation of fungal DNA for PCR. J Clin Microbiol. 2000;38:471.
Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22.
Acknowledgements
We are especially grateful to Dr. Laszlo Majoros from the University of Debrecen, Department of Medical Microbiology for making us possible to test clinical strains from the Debrecen Microbiology Collection. We also thank to Dr. Judit Szabo from the University of Debrecen, Department of Medical Microbiology for performing MALDI-TOF analysis. This study was connected to the project GINOP-2.3.2-15-2016-00042 (Széchenyi 2020 Programme).
Funding
Gábor Fidler received a PhD fellowship from the Medical Science Doctoral Council of the University of Debrecen. The research was supported in part by the GINOP-2.3.2–15–2016-00042 project of the (Széchenyi 2020 Programme) financed by the European Union and the Hungarian Government.
Availability of data and materials
All data generated and analyzed during this study are available within the manuscript file.
Author information
Authors and Affiliations
Contributions
GF performed the construction of the Candida reference and clinical panels conceived the fungal DNA extractions from whole blood samples and the subsequent agarose gel electrophoresis and the real-time PCRs. GF also participated in the statistical analyses. EL and SK performed the cultivation of fungal and bacterial strains and the fungal and bacterial DNA extractions. SK designed the PCR-AFLA method. SB reviewed the manuscript and conducted critical revision of the manuscript for important intellectual content. MP designed the CanTub-simplex PCR method, coordinated the experimental work and also the interpretation of the results. MP drafted the manuscript. All the authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Working with whole blood samples from healthy volunteers was approved by the local ethics committee, MK-JA/50/0096–01/2017. The name of the Ethics committee: Szabolcs-Szatmár-Bereg Megyei Kórházak és Egyetemi Oktatókórház Intézeti Kutatásetikai Bizottság. Adress: 4400 Nyíregyháza, Szent István u. 63., approved by Dr. Diószeghy Péter. The consent was written. All involved patients agreed to participate in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The authors alone are responsible for the content and the writing of the paper.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Intra-, and inter-assay coefficient of variation of the CanTub-simplex PCR on Candida reference panels. Representation of the technical duplicates of the Tm data with mean and ± SD measured on Candida EDTA-WB reference panels at every dilution in a 6-log range. Intra-assay coefficient of variation was estimated for the particular gDNA panels on separarte plates (% C.V.-s of Candida ref-panel_1–7) even as for the plates in a whole (% C.V.-s of ref-plate_1–3). Inter-assay coefficient of variation was estimated for the CanTub-simplex PCR for the reference samples of all panels at every dilution in a 6-log range between three plates finally, grand mean of the sample coefficient of variation (grand % C.V. on ref-plates) was calculated. (DOCX 37 kb)
Additional file 2:
Table S2. Intra-assay coefficient of variation of the CanTub-simplex PCR on 38 Candida clinical strains via EDTA-WB Candida clinical panel. Tm data of Candida clinical strains were grouped according to panels (clin-panel_1–7) and Tm mean was calculated with ±SD. Coefficient of variation (% C.V.) was calculated for every species. Finally, intra- and inter-assay consistencies were calculated. (DOCX 21 kb)
Additional file 3:
Figure S3. CanTub HRM primer alignment results using BLAST algorithm. a) Primer positioning in the Candida albicans beta-tubulin gene with an amplicon size of 203 basepairs. b) Primer positioning in the Candida dubliniensis beta-tubulin gene with an amplicon size of 202 basepairs. c) Primer positioning in the Candida tropicalis beta-tubulin gene with an amplicon size of 204 basepairs. d) Primer positioning in the Candida glabrata chromosome K with an amplicon size of 204 basepairs. (DOCX 38 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fidler, G., Leiter, E., Kocsube, S. et al. Validation of a simplex PCR assay enabling reliable identification of clinically relevant Candida species. BMC Infect Dis 18, 393 (2018). https://doi.org/10.1186/s12879-018-3283-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-018-3283-6