
Abstract
Background
Berry syndrome, a rare combination of cardiac anomalies, consists of aortopulmonary window (APW); aortic origin of the right pulmonary artery; interrupted aortic arch (IAA) or hypoplastic aortic arch or coarctation of the aorta; and an intact ventricular septum. There is lack of review articles that elucidate the clinical features, diagnosis, treatment, and outcomes of Berry syndrome. This publication systematically reviews the 89 cases published since 1982 on Berry syndrome.
Case presentation
A 38-year-old woman presented with a loud murmur and cyanosis. Transthoracic echocardiography demonstrated a severely dilated aorta and main pulmonary artery with a large intervening defect. Distal to the APW, the ascending aorta gave rise to the right pulmonary artery. Additionally, a type A IAA, an intact ventricular septum, and a large patent ductus arteriosus were revealed. Computed tomography angiography with 3-dimensional reconstruction confirmed above findings. This is the first report of a patient of this age with Berry syndrome who did not undergo surgery.
Conclusions
Berry syndrome is a rare but well-identified and surgically correctable anomaly. Patients with Berry syndrome should be followed up for longer periods to better characterize long-term outcomes.
Similar content being viewed by others

Background
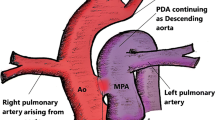
Berry syndrome is characterized by a constellation of abnormalities: aortopulmonary window (APW); aortic origin of the right pulmonary artery (AORPA); interrupted aortic arch (IAA) or hypoplastic aortic arch (HAA) or coarctation of the aorta (CoA); and intact ventricular septum. It is extremely rare, with most cases published as individual reports [1, 2] or in articles describing patients with these abnormalities [3, 4]. Since first described by Berry et al. in 1982 [1], the anomaly has been reported in the English literature in nearly 100 patients. Most were diagnosed in infancy [5, 6], and the surgical procedure was performed immediately. In contrast, our patient was not diagnosed until she was 38 years old. To the best of our knowledge, this is the first report of a patient of this age with Berry syndrome who did not undergo surgery.
We present a review and summary of the English literature available regarding this rare anomaly. A PUBMED-based search was conducted using the primary search terms “Berry syndrome”, “aortopulmonary window”, “aortopulmonary septal defect”, “interrupted aortic arch”, “hypoplastic aortic arch”, “coarctation of aorta”, “aortic origin of right pulmonary artery”, and “hemitruncus”. The flow diagram of the study selection process is shown in Fig. 1. Two additional articles were found by checking reference lists [7, 8]. Four duplicate studies were excluded [9,10,11,12]. Three articles were excluded because the patients did not meet our definition of Berry syndrome [13,14,15]. One article was excluded because the data could not be extracted [16]. Our case, combined with those described in the 48 selected articles, comprised a total of 89 evaluable cases [1,2,3,4,5,6,7,8, 17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56].

Flowchart of the literature selection process
Case presentation
A 38-year-old woman received a tentative diagnosis of congenital cardiac disease in childhood. She is a farmer in Liaoning province (not high altitude) with no special lifestyles. She presented in our outpatient for further diagnosis. Physical examination revealed a loud murmur and cyanosis. Electrocardiogram demonstrated normal rhythm and right ventricular hypertrophy with right axis deviation. Echocardiographic study showed a severely dilated right ventricle, with a diameter of 39 mm; the left ventricle was relatively small, with a diameter of 33 mm. The arterial trunk was markedly enlarged (71–105 mm), with a type III APW (Fig. 2a, b) measuring approximately 65 mm. A right pulmonary artery (RPA) originating from the posterior wall of the ascending aorta was also found, while the origin of the left pulmonary artery (LPA) was normal (Fig. 2a, b). The coronary arteries were normally positioned. Additionally, the patient had a type A IAA (distal to the left subclavian artery), an intact ventricular septum, and a large patent ductus arteriosus (PDA) with bidirectional shunts. The patient had severe pulmonary artery hypertension, approximately equal to the systemic blood pressure, and the left ventricular ejection fraction (LVEF) was nearly 53%. Computed tomography angiography with 3-dimensional reconstruction confirmed the large APW opening into the main and right pulmonary arteries, as well as type A IAA supplied by a large ductal artery (Fig. 2c, d). The patient refused to be hospitalized to further evaluate the possibility of surgery, and she was lost to follow-up. Although missing the opportunity to confirm above findings by surgery, a diagnose of Berry syndrome was made.

Transthoracic echocardiography demonstrated a severely dilated aorta and main pulmonary artery with a large intervening defect (a, b). Distal to the APW, the ascending aorta gave rise to the right pulmonary artery (a, b). Computed tomography showed a type III APW and an AORPA (c). Three-dimensional computed tomography revealed a dilated aortic sac, IAA, and the descending aorta formed via a PDA (d). AORPA, aortic origin of the right pulmonary artery; APW, aortopulmonary window; PDA, patent ductus arteriosus; IAA, interruption of the aortic arch
Discussion and conclusions
Berry syndrome is an extremely rare disease, accounting for only 0.046% of all reported cases of congenital heart anomalies [52]. It is more common in males (67.1%). Patients commonly presented with one or more clinical symptoms, including cyanosis, respiratory distress symptoms, cardiac murmur, symptoms of congestive cardiac failure, and other systemic symptoms (Table 1). The distribution of age at diagnosis in 81 patients was: 13 (16.0%) diagnosed prenatally, 43 (53.1%) diagnosed before 3 months, 13 (16.0%) diagnosed between 3 months and 1 year; 5 (6.2%) diagnosed between 1 and 12 years, and 2 (2.5%) diagnosed after 12 years; 5 of the patients (6.2%) were diagnosed at autopsy.
According to the possible embryogenesis described by Berry et al. [1], when the aortopulmonary septum fails to form, a large APW may represent a partial persistence of the common arterial trunk. Affected by this, the sixth pharyngeal arches may fail to form a bifurcation and may attach improperly to the undivided truncal segment. This results in a wide separation between the right and left pulmonary arteries, and in turn, an AORPA is formed. During fetal development, the APW and AORPA could reduce blood flow in the aortic arch, leading to HAA/CoA/IAA. According to our review, most patients with Berry syndrome have type II APW (78.9%). Consistent with previously reported findings [6], the RPA usually arose from the aorta (84.9%), and most cases presented with type A IAA (88.2%).
Associated cardiac and extracardiac anomalies were common. Among 68 patients for whom such detailed information was available, concomitant cardiac and extracardiac anomalies were diagnosed in 11 (16.2%), including two with an atrial septal defect (one with a small muscular ventricular septal defect and trisomy 13), four with patent foramen ovale, one with abnormalities associated with trisomy 13, one with anomalous origin of the left coronary artery from the main pulmonary artery, one with situs inversus, one with an aberrant right subclavian artery arising from the descending aorta, and one with persistent left superior vena cava. Some authors have reported the important role of neural crest cells in the development of the aortic and pulmonary trunks of the heart [21, 57]. However, the chromosomal abnormalities found in the Berry syndrome cases we reviewed (see Additional file 1) did not seem to be pathogenetically related to a neural crest deformity, which is more likely to be related to a 22q11 gene deletion [57]. To better elucidate the genetic mechanisms underlying the unusual constellation of defects in Berry syndrome, more genetic studies are needed.
Echocardiography is an essential methodology for the diagnosis of complex congenital heart disease. Echocardiography was the initial diagnostic method used in 82.7% of the cases we included. The accuracy rate of echocardiographic diagnosis was 60.9%. In 23.9% of cases, missing recognition of an AORPA resulted in a partially accurate diagnosis. Since AORPA can be readily diagnosed at the time of surgery, it does not increase the difficulty of the surgical procedure or necessitate modification of the therapeutic approach [33]. Thus, echocardiography can provide adequate diagnostic information in most cases, as reported previously [16].
Excluding one patient who was lost to follow-up, the mortality rate for unoperated patients was 100%. The age at death ranged from 1 day to 18 years (median, 1 month). Most unoperated patients died within the first month of life (55.6%). Some of them rapidly developed pulmonary vascular obstructive disease and congestive heart failure [55]. Few unoperated patients may achieve long-term survival, such as the 38-year-old woman we describe in this report. In this case, the arterial trunk was extraordinarily enlarged. On review, we found this distinctive feature in 4 other patients [1, 27, 38, 56], one of whom was not diagnosed until he was 15 years old [1] while other 3 patients underwent surgery before two years old. In Berry syndrome, the arterial trunk has 4 exits (LPA, RPA, PDA, and cranial branches). The LPA and RPA are connected to the lungs. The PDA and cranial branches are connected to the lower and upper body, respectively. Since the PDA was large enough to maintain adequate systemic perfusion to the lower half of the body, a new balance may have been established among the large APW, PDA, and the adaptable ventricles in our case. We speculate that the large arterial trunk may represent a partial persistence of the common arterial trunk and provide a place not only for the full mixing of systemic and pulmonary blood, but also to share volume and pressure overload. This could protect the pulmonary vascular bed and prevent early pulmonary vascular obstructive disease delicately. Thus, these abnormalities (large APW, AORPA, IAA, and large PDA) may have a buffering effect on each other when coexisting with a large arterial trunk, and the affected patients have improved long-term survival. However, our speculation should be confirmed in future studies.
Among all patients with congenital heart disease, 5% to 10% have PDA [58], while almost all of the Berry syndrome patients we included (98.7%) had PDA. The type of PDA was available for 31 cases: 20 patients (64.5%) had a large PDA while 11 patients (35.5%) had constricting PDA. A closed ductus arteriosus or a constricting PDA in a patient with Berry syndrome may predict a poor prognosis. Thus, many patients receive prostaglandin E2 to maintain an open ductus arteriosus until further treatment [8, 47, 51, 53]. On review, more than half of patients diagnosed within 3 months present with a large PDA, while patients diagnosed after 3 months all present with a large PDA. We speculate that the PDA will remain open for a long time in some Berry syndrome patients. In these patients, prophylactic use of prostaglandin may not be necessary before surgery.
The choice of surgical procedure for Berry syndrome remains controversial. One-stage repair is achieved by closing the APW, connecting the RPA to the main pulmonary artery, and establishing aortic arch continuity. Two-stage repair involves placing surgical pulmonary bands to control pulmonary blood flow at the first stage. After the ventilation status improves, the patient undergoes definitive surgical repair [2]. Recently, one-stage surgical repair has been considered to yield more acceptable outcomes [6]. Among the patients we included, 84.1% underwent one-stage repair while others underwent two-stage repair. The age distribution at surgery was: 31 (50.0%) before 1 month, 12 (19.4%) between 1 and 3 months, 14 (22.6%) between 3 months and 1 year, and 4 (6.5%) between 1 and 12 years; only one patient (1.6%) underwent surgery after 12 years. Surgery age ranged from 0.1 to 204.0 months (median, 1.0 months; interquartile range, 0.5–4.0 months). Regardless, there were no differences in mortality within a month postoperatively (x2 = 0, p = 1.0) or survival time within the one-year follow-up period (x2 = 0.43, p = 0.51) between these two groups. For premature and small-for-gestational-age infants, Ghelani et al. [2] advocated two-stage surgery. Premature birth was only reported in four of the Berry syndrome patients we included; of these, 3 underwent two-stage surgery [2, 18, 24], and 1 underwent one-stage surgery [46]. All of these four patients were alive at the times of final follow-up. Given the small sample size and short follow-up period, it is difficult to determine which surgical approach is better.
In pediatric patients younger than three months, the increased pulmonary artery pressure is assumed as mean pulmonary artery pressure exceeded 25 mm Hg [59]. In those older than three months, pulmonary hypertension is defined as mean pulmonary artery pressure exceeded 25 mm Hg [59]. All 19 patients younger than three months, for whom preoperative pulmonary artery pressure was reported, showed increased pulmonary artery pressure. Among 7 patients younger than three months for whom postoperative pulmonary artery pressure was reported, 2 patients (28.6%) showed normal pulmonary artery pressure; 5 patients (71.4%) showed tricuspid regurgitation gradient decreased below 40 mm Hg. All 7 patients older than three months, for whom preoperative pulmonary artery pressure was reported, showed pulmonary hypertension. Among 10 patients older than three months for whom postoperative pulmonary artery pressure was reported, one patient (10%) showed normal pulmonary artery pressure; 9 patients (90%) showed tricuspid regurgitation gradient decreased below 40 mm Hg.
Berry syndrome patients are usually critically ill and need urgent surgery [6]. Because of the large APW and aortic origin of the RPA, most specialists believe that critical pulmonary arterial hypertension appears early after birth [1, 5, 55]. According to some authors, severe pulmonary vascular disease might develop as early as the first 3 months of life due to massive pulmonary blood flow and circulating vasoconstrictors [60]. In our review, almost one-third of the patients were older than 3 months at surgery. With such a delayed treatment, the postoperative pulmonary arterial pressure in most patients was decreased below 40 mmHg [6]. Even in a patient with a right-to-left shunt, the pulmonary artery pressure decreased to the normal range postoperatively [8]. Such outcomes are better than predicted based on the pathophysiology of this complex anomaly.
Among 29 patients for whom information was available, 22 patients (75.9%) experienced postoperative complications during the follow-up period; 20 of 26 patients in the one-stage surgery group (76.9%) had a postoperative complication compared to 2 of 3 (66.7%) in the two-stage surgery group (x2 = 0, p = 1.0). The high rate of postoperative complications indicates the value of long-term follow-up. Complications mainly presented as great vessel narrowing and residual defects. Most patients (58.6%) suffered from RPA stenosis as a result of either compression by the aorta or excessive tension on the pulmonary anastomosis [53]. In the reports we reviewed, 44.8% and 3.4% of the patients had stenosis at the aortic arch and residual APW, respectively, which accords with the findings of a previous study [6]. Reinterventions were required in 13 of 27 patients (48.1%) during the follow-up period: 13 of 25 patients in the one-stage group underwent reinterventions, while neither of the 2 patients in the two-stage surgery group underwent reintervention (x2 = 2.01, p = 0.48). Reinterventions for stenosis at the aortic arch were performed in 9 patients, including surgical repair of residual arch obstruction in 3 and balloon angioplasty of the aortic arch in 6. Reinterventions for RPA stenosis were performed in 7 patients, including surgical repair of RPA obstruction in 5 and balloon angioplasty of the RPA in 3 (one of the patients underwent both surgical repair and balloon angioplasty of the RPA). Analyzed by binary logistic regression, the major morphologic factors (morphology of aortic artery, IAA type, APW type, patent ductus arteriosus type, original RPA site, and type of surgery) were not independent predictors of complications and reintervention after surgery.
In conclusion, Berry syndrome is a rare but well-identified and surgically correctable anomaly. Echocardiography can provide adequate diagnostic information and more than half of the patients diagnosed before 3 months old. Most patients with Berry syndrome presented with type II APW, type A IAA, PDA, and the RPA usually arose from the aorta. Pulmonary hypertension was common in Berry syndrome patients and the postoperative pulmonary arterial pressure usually decreased below 40 mmHg. We found 50.0% patients underwent surgery before 1 month old and no differences in mortality within a month postoperatively or survival time within the one-year follow-up period between different types of surgery. Some patients would experience postoperative complications and reinterventions were required. In addition, we discussed the possible reasons why unoperated patients might achieve long-term survival and whether it is necessary to use prostaglandin prophylactically before surgery. However, the major limitation of this review is that the data analysis was based on the incomplete information available. There is a publication bias since an unknown number of patients died early without a diagnose or being offered surgical repair. Meantime, long-term survivors included in cohort studies describing patients with cardiac anomalies (e.g. IAA, APW, AORPA and PDA) may not be recognized in our literature search. Therefore, the results of this review should be treated with caution. Patients with Berry syndrome should be followed up for longer periods to better characterize long-term outcomes.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its Additional file 1].
Abbreviations
- APW:
-
Aortopulmonary window
- AORPA:
-
Aortic origin of the right pulmonary artery
- IAA:
-
Interrupted aortic arch
- HAA:
-
Hypoplastic aortic arch
- CoA:
-
Coarctation of the aorta
- RPA:
-
Right pulmonary artery
- PDA:
-
Patent ductus arteriosus
- LPA:
-
Left pulmonary artery
References
Berry TE, Bharati S, Muster AJ, Idriss FS, Santucci B, Lev M, et al. Distal aortopulmonary septal defect, aortic origin of the right pulmonary artery, intact ventricular septum, patent ductus arteriosus and hypoplasia of the aortic isthmus: a newly recognized syndrome. Am J Cardiol. 1982;49(1):108–16.
Ghelani SJ, Quinonez LG, Rathod RH. Prenatal diagnosis and management of berry syndrome, a rare conotruncal anatomy. Circulation. 2015;132(16):1593–4.
Backer CL, Mavroudis C. Surgical management of aortopulmonary window: a 40-year experience. Eur J Cardiothorac Surg. 2002;21(5):773–9.
Liu Y, Cheng L, Qian X, Zhu H, Duan W, Yu S, et al. Surgical correction of anomalous origin of one pulmonary artery without grafts in infants. J Card Surg. 2015;30(1):85–91.
Sharma J, Saleh M, Das BB. Berry syndrome with trisomy 13. Pediatr Cardiol. 2002;23(2):205–9.
Hu R, Zhang W, Liu X, Dong W, Zhu H, Zhang H. Current outcomes of one-stage surgical correction for Berry syndrome. J Thorac Cardiovasc Surg. 2017;153(5):1139–47.
Abdulrahman R, Samadi M, Negargar S. A neonate with Berry syndrome (AP window with interrupted aortic arch). J Cardiovasc Thorac Res. 2009;1(3):51–3.
Razzaq S, Aslam N, Ahmad W, Amanullah M. Berry syndrome: one stage surgical repair in a neonate. Int J Case Rep Images. 2017;8(12):770–5.
Chiu IS, Wu SJ, Lee ML. One-stage repair of interrupted aortic arch and aortopulmonary window with an autologous arterial flap. J Card Surg. 1999;14(4):306–9.
Erdem A, Aydemir NA, Demir H, Zeybek C, Saritas T, Akdeniz C, et al. Anomalous origin of one pulmonary artery branch from the ascending aorta: experience of our center. Turk Kardiyol Dern Ars. 2010;38(6):411–5.
Takeuchi K, Masuzawa A, Kobayashi J, Yoda H. One-stage repair of interrupted aortic arch in combination with aortopulmonary window–preoperative three-dimensional computed tomography. Eur J Cardiothorac Surg. 2011;39(1):136.
Wu J, Geng B, Wan X, Li W, Zhang G. Prenatal diagnosis of berry syndrome by fetal echocardiography. J Ultrasound Med. 2016;35(4):848–9.
Alsoufi B, Schlosser B, McCracken C, Kogon B, Kanter K, Border W, et al. Current outcomes of surgical management of aortopulmonary window and associated cardiac lesions. Ann Thorac Surg. 2016;102(2):608–14.
Talwar S, Agarwal P, Choudhary SK, Kothari SS, Juneja R, Saxena A, et al. Aortopulmonary window: morphology, diagnosis, and long-term results. J Card Surg. 2017;32(2):138–44.
Shahdadpuri R, Prendiville T, Nolke L, McMahon CJ. Berry syndrome in association with familial limb malformation. Ir Med J. 2009;102(2):54–6.
Li J, Yang Y, Duan X, Jin L, Zheng L, Zhang X, et al. Berry syndrome: a rare cardiac malformation with extra-cardiac findings. Sci China Life Sci. 2017;60(7):772–4.
Braunlin E, Peoples WM, Freedom RM, Fyler DC, Goldblatt A, Edwards JE. Interruption of the aortic arch with aorticopulmonary septal defect. An anatomic review Pediatr Cardiol. 1982;3(4):329–35.
Tabak C, Moskowitz W, Wagner H, Weinberg P, Edmunds LH Jr. Aortopulmonary window and aortic isthmic hypoplasia. Operative management in newborn infants. J Thorac Cardiovasc Surg. 1983;86(2):273–9.
Pettersson G, Sabel KG, Sudow G. Total anatomic correction of interrupted aortic arch complex. Experience in 4 infants. Scand J Thorac Cardiovasc Surg. 1986;20(1):5–10.
Mendoza DA, Ueda T, Nishioka K, Yokota Y, Mikawa H, Nomoto S, et al. Aortopulmonary window, aortic origin of the right pulmonary artery, and interrupted aortic arch: detection by two-dimensional and color Doppler echocardiography in an infant. Pediatr Cardiol. 1986;7(1):49–52.
Kutsche LM, Van Mierop LH. Anatomy and pathogenesis of aorticopulmonary septal defect. Am J Cardiol. 1987;59(5):443–7.
Kutsche LM, Van Mierop LH. Anomalous origin of a pulmonary artery from the ascending aorta: associated anomalies and pathogenesis. Am J Cardiol. 1988;61(10):850–6.
Ding WX, Su ZK, Cao DF, Jonas RA. One-stage repair of absence of the aortopulmonary septum and interrupted aortic arch. Ann Thorac Surg. 1990;49(4):664–6.
Yoo SJ, Choi HY, Park IS, Hong CY, Song MG, Kim SH. Distal aortopulmonary window with aortic origin of the right pulmonary artery and interruption of the aortic arch (Berry syndrome): diagnosis by MR imaging. AJR Am J Roentgenol. 1991;157(4):835–6.
Sreeram N, Walsh K. Aortopulmonary window with aortic origin of the right pulmonary artery. Int J Cardiol. 1991;31(2):249–51.
Boonstra PW, Talsma M, Ebels T. Interruption of the aortic arch, distal aortopulmonary window, arterial duct and aortic origin of the right pulmonary artery in a neonate: report of a case successfully repaired in a one-stage operation. Int J Cardiol. 1992;34(1):108–10.
Chiu IS, Wang JK, Wang MJ, Wang CC. One-stage repair of aortopulmonary septal defect and interrupted aortic arch. Ann Thorac Surg. 1994;58(5):1529–32.
Burke RP, Rosenfeld HM. Primary repair of aortopulmonary septal defect, interrupted aortic arch, and anomalous origin of the right pulmonary artery. Ann Thorac Surg. 1994;58(2):543–5.
Alva-Espinosa C, Jimenez-Arteaga S, Diaz-Diaz E, Martinez-Sanchez A, Jimenez-Zepeda D, Mojarro-Rios J, et al. Diagnosis of Berry syndrome in an infant by two-dimensional and color Doppler echocardiography. Pediatr Cardiol. 1995;16(1):42–4.
Kim TK, Choe YH, Kim HS, Ko JK, Lee YT, Lee HJ, et al. Anomalous origin of the right pulmonary artery from the ascending aorta: diagnosis by magnetic resonance imaging. Cardiovasc Intervent Radiol. 1995;18(2):118–21.
Sharma R, Saha K, Kothari SS. Neonatal correction of interrupted aortic arch, aortopulmonary window and ascending aortic origin of right pulmonary artery. Indian Heart J. 1996;48(6):717–20.
Abbruzzese PA, Merlo M, Chiappa E, Bianco R, Ferrero F, Cappone CM. Berry syndrome, a complex aortopulmonary malformation: one-stage repair in a neonate. Ann Thorac Surg. 1997;64(4):1167–9.
Carrel T, Pfammatter JP. Interrupted aortic arch, aorto-pulmonary window and aortic origin of the right pulmonary artery: single stage repair in a neonate. Eur J Cardiothorac Surg. 1997;12(4):668–70.
Codispoti M, Mankad PS. One-stage repair of interrupted aortic arch, aortopulmonary window, and anomalous origin of right pulmonary artery with autologous tissues. Ann Thorac Surg. 1998;66(1):264–7.
Lee ML. Recognition of Berry syndrome in a 4-day-old neonate by echocardiography and transvenous angiocardiography. Int J Cardiol. 1999;71(1):93–5.
Senzaki H, Asano H, Masutani S, Matunaga T, Ishido H, Taketatu M, et al. Anomalous origin of the left coronary artery from the main pulmonary artery associated with Berry syndrome. J Thorac Cardiovasc Surg. 2003;126(5):1645–7.
Fong NC, Kong CT, Mak WY, Shiu YK, Lee SY, Chow CB, et al. Early detection of Berry syndrome in a newborn with differential cyanosis. Chin Med J (Engl). 2006;119(17):1485–8.
Chen CA, Chiu SN, Wu ET, Lin MT, Wang JK, Chang CI, et al. Surgical outcome of aortopulmonary window repair in early infancy. J Formos Med Assoc. 2006;105(10):813–20.
Choo KS, Lee HD, Ban JE, Sung SC, Chang YH, Kim CW, et al. Evaluation of obstructive airway lesions in complex congenital heart disease using composite volume-rendered images from multislice CT. Pediatr Radiol. 2006;36(3):219–23.
Konstantinov IE, Karamlou T, Williams WG, Quaegebeur JM, del Nido PJ, Spray TL, et al. Surgical management of aortopulmonary window associated with interrupted aortic arch: a Congenital Heart Surgeons Society study. J Thorac Cardiovasc Surg. 2006;131(5):1136–41.
Murayama H, Watanabe T, Yasuda K, Kobayashi A. Mirror image Berry syndrome: a case report of complex aortopulmonary malformation in viscero-atrial situs inversus. Minerva Chir. 2007;62(2):129–32.
Park SY, Joo HC, Youn YN, Park YH, Park HK. Berry syndrome: two cases of successful surgical repair. Circ J. 2008;72(3):492–5.
Hayashi G, Inamura N, Kayatani F, Kawazu Y, Hamamichi Y. Prenatal diagnosis of aortopulmonary window with interrupted aortic arch by fetal echocardiography. Fetal Diagn Ther. 2010;27(2):97–100.
Matsubara Y, Ota M, Bito A, Katayama T, Matsubara K, Ito M. Prenatal diagnosis of Berry syndrome by fetal echocardiography. Ultrasound Obstet Gynecol. 2010;35(3):374–6.
Morito H, Masuzawa A, Kobayashi J, Takeuchi K. One-Stage Surgical Repair for Berry Syndrome With Preoperative Diagnosis by 3-Dimensional CT. World J Pediatr Congenit Heart Surg. 2011;2(3):491–4.
Mannelli L, Mosca R, Henry G, Srichai-Parsia MB. A case of berry syndrome type 2B. Congenit Heart Dis. 2011;6(4):389–92.
Jayaram N, Knowlton J, Shah S, Gelatt M, Lofland G, Raghuveer G. Berry syndrome: a possible genetic link. Pediatr Cardiol. 2013;34(6):1511–3.
Demir IH, Erdem A, Saritas T, Demir F, Erol N, Yucel IK, et al. Diagnosis, treatment and outcomes of patients with aortopulmonary window. Balkan Med J. 2013;30(2):191–6.
Wang J, Song Y, Cheng TO, Xie M, Wang X, Yuan L, et al. The value of transthoracic echocardiography in the diagnosis of anomalous origin of the right pulmonary artery from the ascending aorta: A single center experience from China. Int J Cardiol. 2015;184:750–4.
Yang SH, Tian XX, Li YY, Yang ZJ. Prenatal diagnosis of Berry syndrome by fetal echocardiography: a case report. Echocardiography. 2016;33(10):1611–3.
Remon JI, Briston DA, Stern KW. Berry syndrome: the importance of genetic evaluation before surgical intervention. Cardiol Young. 2016;26(1):188–90.
Zhang X, Liu XW, Gu XY, Han JC, Hao XY, Fu YW, et al. Prenatal diagnosis of Berry syndrome by fetal echocardiography: A report of four cases. Echocardiography. 2018;35(4):563–5.
Binsalamah ZM, Greenleaf CE, Heinle JS. Type A interrupted aortic arch and type III aortopulmonary window with anomalous origin of the right pulmonary artery from the aorta. J Card Surg. 2018;33(6):344–7.
Yu S, Han J, Gao S, Liu X, Gu X, Zhang Y, et al. The prenatal diagnosis of aortopulmonary window by fetal echocardiography. Echocardiography. 2018;35(11):1835–40.
Li W, Bin G, Jiang W, Shuang Y. Prenatal diagnosis of aortopulmonary window by 2-dimensional echocardiography: summary of 8 cases. J Ultrasound Med. 2019;38(3):795–803.
Bu H, Zhao T. Berry syndrome diagnosed by three-dimensional computed tomographic angiography. Acta Cardiol. 2020;75(2):160–1.
Bergwerff M, Verberne ME, DeRuiter MC, Poelmann RE, Gittenberger-de Groot AC. Neural crest cell contribution to the developing circulatory system: implications for vascular morphology? Circ Res. 1998;82(2):221–31.
Schneider DJ, Moore JW. Patent ductus arteriosus. Circulation. 2006;114(17):1873–82.
Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation. 2015;132(21):2037–99.
Prifti E, Bonacchi M, Murzi B, Crucean A, Bernabei M, Luisi VS, et al. Anomalous origin of the left pulmonary artery from the aorta. Our experience and literature review. Heart Vessels. 2003;18(2):79–84.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81571686). The funder had no role in the study design, data collection, analysis, interpretation of data, and preparation of the manuscript or decision to publish.
Author information
Authors and Affiliations
Contributions
WB and WR designed the study. WB drafted the manuscript. WB, YX, YL, YH and WR significantly contributed to the imaging diagnosis, literature search, data extraction or data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethical Committee of Shengjing Hospital approved this work (2019PS524K).
Consent for publication
Written informed consent for publication of the clinical details and images was obtained from the patient. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Literature review of 89 cases of Berry syndrome from 1982 to June 2019. The following clinical data were extracted and reviewed: sex, presence of clinical symptoms, premature birth, age at diagnosis, diagnostic methods, anomalies recognized by echocardiography, anatomic features of the cardiac anomalies (morphology of aortic artery, APW type, type of ductus arteriosus, and original site of RPA), associated cardiac and extracardiac anomalies, chromosomal abnormalities, age at surgery, types of surgery, postoperative details (postoperative hospitalization, postoperative follow-up time, postoperative complications, and reintervention after surgery), outcome, cause of death, preoperative and postoperative pulmonary artery pressure, and the presence of a massively dilated vascular sac.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bi, Wj., Xiao, Yj., Liu, Yj. et al. Berry syndrome: a case report and literature review. BMC Cardiovasc Disord 21, 15 (2021). https://doi.org/10.1186/s12872-020-01837-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-020-01837-y