
Abstract
Background
Although most cases of atrial septal defect (ASD) are sporadic, familial cases have been reported, which may be caused by mutation of transcription factor GATA binding protein 4 (GATA4). Herein we combined whole-exome sequencing and bioinformatics strategies to identify a novel mutation in GATA4 accounting for the etiology in a Chinese family with ASD.
Methods
We identified kindred spanning 3 generations in which 3 of 12 (25.0%) individuals had ASD. Punctilious records for the subjects included complete physical examination, transthoracic echocardiography, electrocardiograph and surgical confirming. Whole-exome capture and high-throughput sequencing were performed on the proband III.1. Sanger sequencing was used to validate the candidate variants, and segregation analyses were performed in the family members.
Results
Direct sequencing of GATA4 from the genomic DNA of family members identified a T-to-C transition at nucleotide 929 in exon 5 that predicted a methionine to threonine substitution at codon 310 (M310T) in the nuclear localization signal (NLS) region. Two affected members (II.2 and III.3) and the proband (III.1) who was recognized as a carrier exhibited this mutation, whereas the other unaffected family members or control individuals did not. More importantly, the mutation GATA4 (c.T929C: p.M310T) has not been reported previously in either familial or sporadic cases of congenital heart defects (CHD).
Conclusions
We identified for the first time a novel M310T mutation in the GATA4 gene that is located in the NLS region and leads to family ASD with arrhythmias. However, the mechanism by which this pathogenic mutation contributes to the development of heart defect and tachyarrhythmias remains to be ascertained.
Similar content being viewed by others


Introduction
Atrial septal defect (ASD) is a common cardiovascular malformation, accounting for 10% of congenital heart defects (CHD), which is one of the major birth defects in the world. ASD is often reported in sporadic form; however, the reported familial cases have more research value [1,2,3]. ASD may be isolated or associated with other CHDs, such as pulmonary valve stenosis (PVS), ventricular septal defect (VSD) and conduction defects, one of the study found that GATA4 genetic variations are associated with ASD, TOF and VSD in South Indian patients. In silico analysis provides further evidence that some of the observed mutations are pathogenic [4]. In addition, persistent left to right blood shunt may result in atrial dysfunctions and atrial arrhythmias, in the absence of surgical or catheter-based repair [5]. Therefore, CHD is still a serious threat to human, so the early prenatal screening and diagnosis for this type of birth defect are urgently required.
Although the CHD etiology is too complicated to be well characterized because of the complexity of heart development, numerous intrinsic factors [6] (genetic factors) and extrinsic threats [7] (environmental) were identified as contribution to CHD. Many candidate genes such as GATA4, TBX5, NKX2.5, BMP4 and HAND1 have been proven to be responsible for heart development and diseases [8]. GATA4 is one of the most widely investigated genes in CHD, with over 100 known mutation sites, which are related to the structural heart defect such as ASD, VSD, and PVS [9]. Recently, the phenotypic genetics of familial ASD has been widely investigated, and transcription factors as an important mediator in cardiac development are still the focus of attention [10,11,12]. Intrinsic factors have been identified as a major contributor to the pathogenesis of family ASD with the development of sequencing technology [13], and mutations in the GATA4 gene have been identified as a pathogenic factor of familial ASD [3, 14].
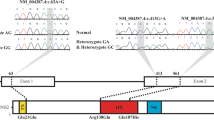
The zinc-finger transcription factors were encoded by GATA binding protein 4 (GATA4), which is essential for heart development [15, 16] and considered to be a gene regulating embryogenesis and myocardial differentiation and function, and bound the GATA motif which is present in the promoters of many genes [17]. GATA4 has 442 amino acids, including the N-terminus zinc fingers (NZf), the C-terminus zinc fingers (CZf) and the nuclear localization signal (NLS) [2]. More importantly, the 271–322 amino acid fragment in the DNA binding domain has been reported and proved to be the smallest functional NLS region, which is vital to the process of cardiac development [18]. In the current study, we checked out a clinically characterized family with a diagnosis of ASD. We found an obvious autosomal-dominant inheritance with reduced penetrance in this family. In addition, after performing surgical confirming and surgical repairs on patients, we conducted a clinical and genetic analysis and identified for the first time a novel pathogenic mutation of GATA4 in the NLS region (NM_002052: exon5: c.T929C: p.M310T) by whole-exome sequencing of the patient in the family, which was confirmed by Sanger sequencing. Taken together, our study strongly suggests that the dominant family ASD involved in this study may be caused by GATA4 gene deficiency.
Patients and methods
Patients and clinical examination
The present study enrolled 3 patients (Fig. 1a) with ASD and arrhythmia from The Second Xiangya Hospital of Central South University (Changsha, China). The study group comprised of 5 male and 7 female patients (Table 1). Punctilious records for the subjects included a complete physical examination, a transthoracic echocardiography, a 12-lead electrocardiograph and a surgical confirming. All noninvasive exam results are confirmed in the surgery.

a Pedigree of the affected family is comprised of three generations. The squares and circles indicate males and females, respectively. Participating members of each generation are indicated numerically. The arrow appoints the proband of the family. The mutation, c.T929C in GATA4, has been demonstrated that segregated in this family; b All members with the heart defects were diagnosed by transthoracic echocardiography. ASD is clearly displayed through 3D reconstruction (bottom). ASD, atrial septal defect
The study protocol was approved by Review Board of the Second Xiangya Hospital of Central South University (Changsha, China). Written informed consents for the publication of the patient’s information were obtained from the parents of the patient and patents themselves.
DNA extraction
Genomic DNA was extracted from peripheral blood lymphocytes of each patient and the family members. Genomic DNA was prepared for testing with a DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA) on a QIA cube automated DNA-extraction robot (Qiagen, Hilden, Germany) [19]. The quality and quantity of the DNA samples were measured by a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA), after which 2 µl DNA from each sample was used for analysis [20, 21].
Whole-exome sequencing (WES) and filtering
At the Novogene Bioinformatics Institute (Beijing, China), we used whole-exome capture and high-throughput sequencing (HTS) technology to detect the proband III.1 systematically. In short, the Agilent SureSelect Human All ExonV5 Kit (Agilent, California, USA) was purchased and used to capture the whole exomes, which were then sequenced on the Illumina HiSeq. 2500 platform [22]. Consistent with the human reference genome (UCSC hg19), the details of the sequencing data are shown in Fig. 2. The following criteria were used as filtering criteria for single nucleotide variants (SNVs) and short InDels [23]: (1) synonymous mutations and variants, such as intergenic variation, intronic variation, and UTR regions variation, should be excluded in subsequent analysis; (2) High-frequency (minor allele frequency > 0.01) polymorphisms should be excluded from the databases, such as 1000 Genomes Project, ExAC, ESP6500, and Novogene Bioinformatics Institute internal Exome Sequencing databases; (3) According to the identification principle of new pathogenic genes, the known pathogenic genes should be excluded, and then the variations of 42 known CHD-related genes [24] were listed. Any compound heterozygotes of known genes can be found by this method. (4) Runs of homozygosity (ROH) analysis [22], is a vital method because it can effectively eliminate false-positive variation in the situation of a large number of deletion on the other allele, should be performed due to the examination of consanguineous families. Refer to Fig. 2 for detailed filtering steps.

Schematic representation of filtering strategies applied in this research. The filtering process was applied according to several strategies that are demonstrated in the schematic representation
Bioinformatics analysis
Bioinformatics programs, such as MutationTaster, Sift, PROVEAN, Polyphen-2, and LRT, were used to predict the effects of identified variants on protein function, and the Swiss model tools were used to determine the potential effects on protein structure. To further assess the protective effect of the identified variants, we obtained and compared the amino acid sequences of target genes in different species from MutationTaster (http://www.mutationtaster.org), and then the domain analyses were carried out in NCBI Conserved Domains websites (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).
Mutation validation and co-segregation analysis
Sanger sequencing in forward and reverse directions was used to validate the candidate variants identified by WES, and segregation analyses were performed in the family members. Primers pairs used to amplify fragments encompassing individual variants were designed using an online tool (Primer Quest, IDT) (http://www.idtdna.com/Primerquest/Home/Index), and the sequences of the primers as follows: Forward primer: 5′-TCTTTCTCGCTGAGTTCC-3′, Reverse primer: 5′-TTGAGTTGAGCCTGCTTC-3'.
Results
Clinical features
We identified 3 patients, the father and two of the daughters, with multiple complex phenotypes including CHD and arrhythmia in a Chinese family (Fig. 1a). The proband III.1, a 14-year-old patient from Hunan province of Central-South China, was diagnosed as secundum ASD with atrial septal aneurysm, partial atrioventricular septal defect (PAVSD), PVS and persistent left superior vena cava by transthoracic echocardiography (Fig. 1b) and sinus tachycardia, frequent junctional premature beat with aberrant ventricular conduction by HOLTER.
The father II.2 was diagnosed as secundum ASD, PS and ascending aortic dilatation by transthoracic echocardiography (Fig. 1b) and atrial fibrillation paroxysmal ventricular tachycardia by HOLTER. The other patient III.3 was diagnosed as secundum ASD, mild PS and persistent left superior vena cava by transthoracic echocardiography (Fig. 1b) and sinus arrhythmia (sinus tachycardia alternating with sinus bradycardia), cardiac rhythm migration between atrionector and junction by HOLTER. Other members of the family did not show any structural heart defect or severe arrhythmia in the examination. All structural defects were confirmed in the operation. Proband III.1 presented ventricular tachycardia after sternotomy and the heart was irritable in the whole procedure. All clinical details have been reported in Table 1.
Genetic analysis
WES was performed on proband III.1 and the sequence read of 4.7Gbp was generated. The mean depth of the target region is 47.3 × and 95.7% of the targeted bases were covered sufficiently to pass the threshold for SNVs and InDels (Additional file 1: Table S1). Several filtering methods were performed to analyze the known SNVs and InDels. After alignment and SNV calling, 51,328 variants were detected in the proband’s exome. We used several databases to exclude all exonic InDels, non-synonymous variants, and nonsense and splice-site SNVs. Then we ranked genes by Sift, Polyphen-2, and Mutation Taster, 11 variants were identified and prioritized them by patients’ phenotype. 42 CHD-related genes were used to cross-contrast the 11 variants. Eventually, only the mutation GATA4 NM_002052: exon5: c.T929C: p.M310T could be confirmed in the other two affected family members (II.2 III.3) and could not be detected in other healthy parent or normal control.
M310T mutation was confirmed by Sanger sequencing from the samples of available members of the affected family (Fig. 3). At the same time, several bioinformatics programs (SIFT, Polyphen-2, Mutation Taster, PROVEAN, and LRT) were used to confirm the pathogenicity of the mutation of M310T, including the physical and chemical characteristics of amino acids at mutation sites, sequence conservation of mutation sites and their adjacent regions, protein structure characteristics, evolution characteristics, etc. The GATA4 (c.T929C, p.M310T) gene mutation was predicted to be ‘Disease-causing’, ‘possibly damaging’, ‘Damaging’, ‘Deleterious’, and ‘Deleterious’ by Mutation Taster (Fig. 4a), PolyPhen-2 (Fig. 4b), SIFT (Fig. 4c) and LRT, respectively. In addition, compared with the wild-type (Fig. 4d), the potential effect of the mutant (Fig. 4e) on the protein structure was determined using the Swiss Model tools. Finally, we submitted the novel single nucleotide polymorphisms (SNPs) to the dbSNP database and received a submitted SNP (ss) number (2,137,544,112).

All members with the secundum atrial septal defect had a GATA4 mutation (left), a T-to-C transition at nucleotide 929 in exon 5 of GATA4. Other unaffected family members with wild type alleles of GATA4 are shown (right); Wild type (WT) and mutated (MT) amino acid sequences of GATA4 protein (bottom). The T929C transition creates a methionine to threonine substitution at codon 310

Results of several bioinformatics programs for the pathogenicity of the mutation of M310T. Results of Mutation Taster (a), Polymorphism Phenotyping v2 predictions (b), and Sorting Intolerant From Tolerant (c); The protein structure was determined using the Swiss Model tools (d, e)
Discussion
ASD is the third most common type of congenital heart disease, of which about 65–70% are secundum defects [25]. In these patients, changes in cardiac structure are triggered by increased blood flow resulting from a left-to-right shunt due to intracardiac defect. Therefore, early detection and surgical treatment are the main strategies. Thanks to the development of technique, including the introduction of cardiopulmonary bypass and accompanying large-scale heart surgery skills improvement, the mortality of ASD has been dramatically reduced [26]. However, due to the complexity of heart development, the exact causes of ASD, especially for the complex overlapping phenotype of ASD, still need more ongoing research efforts though we know that knowledge of heart development and function is the absolute necessity for better survival of human. Nowadays, the discovery of genetic causes of ASD has been being accelerated by many new technologies including single nucleotide polymorphism arrays, next-generation sequencing (NGS), and copy number variant platforms [27]. Among all new genetic technologies, the application of NGS in various aspects of heart biology has resulted in discoveries, generating novel insights into this field of study [28]. In the present study, WES combined ROH was employed to find out the suspicious disease-causing gene in one consanguineous family. This technique has been considered as a rapid and cost-effective tool for screening the new variants or genes for rare Mendelian unknown disorders [29, 30]. It helps genetic diagnostics for clinical cases with a mutational spectrum of known and unknown diseases. Some filtering strategies are needed for excluding variants that are implausible to cause disease because sometimes it is difficult to identify between pathogenic and benign mutations in the WES results [31]. With the analysis of WES result in this study, we quickly determined the most possible pathogenic mutation in this family is GATA4 p.M310T and confirmed the result by Sanger sequencing.
GATA4 gene belongs to a GATA family, which is consisted of 6 structure-conserved transcription factors. GATA4 gene, which is expressed in the cardiac system and endodermal derivatives [32], is a highly conserved transcriptional factor with seven exons. The GATA4 protein is comprised of TAD, NZF and NLS [2, 33]. Many shreds of evidence showed that GATA4 plays significant roles in many stages of heart development, including looping morphogenesis, septation, ventricular myocardium proliferation, and heart contraction [34]. For this reason, GATA4 was considered as a regular candidate for CHD genetic screening. Many mutations in the GATA4 coding region have been identified as the genotype of CHD patients and not all of them were predicted by bioinformatics tools, such as PROVEAN and SIFT, as the pathogenic genes [34]. NLS was considered as a crucial role in ASD epidemiology because 5 family cases were reported in this region, include S52F, G296S, 1074 (delC), 1075 (delG) and M310V [2, 3, 35]. Especially in our present study, we determined a different protein mutation in the same protein location (M310V) as reported [2]. What’s more, in the mouse mutation study, M310V transgenic mice had shown a higher incidence of CHD than wild-type control mice [36], which indicated codon 310 in the GATA4 gene is a CHD-related pathogenic coding region.
Here, we report for the first time an M310T mutation in the NLS region, which is necessary and sufficient for GATA4 transcription factor activity and cardiac development. Using the Swiss model tool, we identified the NLS region as the potential impact region of the mutation on protein structure (Fig. 4). The changes in this region may lead to a decrease of transcriptional activity, thus affecting the transcriptional activation process during development. What's more, the region affected by M310T mutation is also immediately adjacent to CZf region, which is crucial for DNA binding and cofactor interaction [36, 37]. In addition, Garg reported [14] that the G296S mutation disrupts the DNA-binding and transactivation activity of GATA4 and destroys the synergy in transcriptional activation between GATA4 and its cofactor TBX5, resulting in heart anomalies such as pulmonary stenosis, atrioventricular septal defect, and ASD. Therefore, GATA4 mutation in NLS region may also affect the expression of other transcription factors (TBX5 or NKX2.5) [14, 35], which are crucial in the development of heart, resulting in the observation of ASD in three affected members of this family.
ASD families mostly present the same subtype structural defects without arrhythmia. In this study, all affected family members presented similar defects with distinct differences and all 3 patients were detected different tachyarrhythmia. Although some reported arrhythmias and conduction disorders may be associated with atrial septal defects [38], the reason is unknown and some transcriptional factors genes mutations were related to the ASD family with arrhythmias [39], one possible evidence shown by computationally Mattapally et al. [40], where they established that NKX2.5 cooperativity with GATA4 facilitates its activating and repressing functions [41,42,43]. The interaction between NKX2.5 (TN domain) and GATA4 might also be important for the function as a repressor of ion channels and its downstream target genes. Therefore, they speculate that mutation present in TN domain of NKX2.5 gene will result loss of NKX2.5 and GATA4 interaction, thus will lead to loss of several activator and repressor function of this complex. GATA4 p.M310T in future studies we also need show any effect activator and repressor function have trachyarrythemia. Further investigation is needed for the different phenotypes with the same genotype in the ASD family with arrhythmias.
CHD is a heavy load for the young family and the whole society. In China, the prevalence is still high although many CHD fetuses were aborted when the prenatal screening predicted highly possible of CHD. In the past two decades, cardiac development and genetic studies had provided much detailed information and identified many critical genes in the development of the heart. Although many mutants of these genes had been screened out, more biochemical methods and vivo models should be adopted for confirming. A limitation of this study is that only genetic screening methods had been employed but no furthermore study, such as vivo models, were used. Further studies will be conducted in the future to study the pathogenic mechanism of ASD families with arrhythmias and the reason for different phenotypes with the same genotype.
Conclusion
We identified for the first time a novel M310T mutation in the GATA4 gene that is located in the NLS region and leads to family ASD with arrhythmias. However, the mechanism by which this pathogenic mutation contributes to the development of heart defect and tachyarrhythmia remains to be ascertained.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the dbSNP database repository, persistent web link: https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ss.cgi?subsnp_id=ss2137544112.
Abbreviations
- ASD:
-
Atrial septal defect
- CHD:
-
Congenital heart defects
- PVS:
-
Pulmonary valve stenosis
- VSD:
-
Ventricular septal defect
- PAVSD:
-
Partial atrioventricular septal defect
- TAD:
-
Transactivation domains
- NZf:
-
N-terminus zinc fingers
- CZf:
-
C-terminus zinc fingers
- NLS:
-
Nuclear localization signal
- WES:
-
Whole-exome sequencing
- HTS:
-
High-throughput sequencing
- SNVs:
-
Single nucleotide variants
- ROH:
-
Runs of homozygosity
- SNPs:
-
Single nucleotide polymorphisms
References
Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–900.
Chen Y, Han ZQ, Yan WD, Tang CZ, Xie JY, Chen H, Hu DY. A novel mutation in GATA4 gene associated with dominant inherited familial atrial septal defect. J Thorac Cardiovasc Surg. 2010;140(3):684–7.
Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N, et al. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet. 2004;41(7):e97.
Mattapally S, Nizamuddin S, Murthy KS, Thangaraj K, Banerjee SK. c.620C>T mutation in GATA4 is associated with congenital heart disease in South India. BMC Med Genet. 2015;16:7.
Sarkozy A, Conti E, Neri C, D’Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 2005;42(2):e16.
Bentham J, Bhattacharya S. Genetic mechanisms controlling cardiovascular development. Ann N Y Acad Sci. 2008;1123:10–9.
Wang J, Cao H, Sun D, Qi Z, Guo C, Peng W, Sun Y, Xie Y, Liu X, Li B, et al. Associations between ambient air pollution and mortality from all causes, pneumonia, and congenital heart diseases among children aged under 5 years in Beijing, China: a population-based time series study. Environ Res. 2019;176:108531.
Paige SL, Plonowska K, Xu A, Wu SM. Molecular regulation of cardiomyocyte differentiation. Circ Res. 2015;116(2):341–53.
Su W, Zhu P, Wang R, Wu Q, Wang M, Zhang X, Mei L, Tang J, Kumar M, Wang X, et al. Congenital heart diseases and their association with the variant distribution features on susceptibility genes. Clin Genet. 2017;91(3):349–54.
Olson EN, Srivastava D. Molecular pathways controlling heart development. Science. 1996;272(5262):671–6.
Olson EN. A genetic blueprint for growth and development of the heart. Harvey Lect. 2002;98:41–64.
Fishman MC, Olson EN. Parsing the heart: genetic modules for organ assembly. Cell. 1997;91(2):153–6.
Nemer M. Genetic insights into normal and abnormal heart development. Cardiovasc Pathol. 2008;17(1):48–54.
Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443–7.
Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11(8):1061–72.
Gajewski K, Fossett N, Molkentin JD, Schulz RA. The zinc finger proteins Pannier and GATA4 function as cardiogenic factors in Drosophila. Development. 1999;126(24):5679–88.
Kobayashi M, Nishikawa K, Yamamoto M. Hematopoietic regulatory domain of gata1 gene is positively regulated by GATA1 protein in zebrafish embryos. Development. 2001;128(12):2341–50.
Philips AS, Kwok JC, Chong BH. Analysis of the signals and mechanisms mediating nuclear trafficking of GATA-4. Loss of DNA binding is associated with localization in intranuclear speckles. J Biol Chem. 2007;282(34):24915–27.
Liu L, Bu H, Yang Y, Tan Z, Zhang F, Hu S, Zhao T. A targeted, next-generation genetic sequencing study on tetralogy of fallot, combined with cleft lip and palate. J Craniofac Surg. 2017;28(4):e351–5.
Bu H, Liu L, Hu S, Tan Z, Zhao T. Targeted nextgeneration sequencing for research and diagnostics in congenital heart disease, and cleft lip and/or palate. Mol Med Rep. 2019;19(5):3831–40.
Tan ZP, Xie L, Deng Y, Chen JL, Zhang WZ, Wang J, Yang JF, Yang YF. Whole-exome sequencing identifies Y1495X of SCN5A to be associated with familial conduction disease and sudden death. Sci Rep. 2014;4:5616.
Guo T, Tan ZP, Chen HM, Zheng DY, Liu L, Huang XG, Chen P, Luo H, Yang YF. An effective combination of whole-exome sequencing and runs of homozygosity for the diagnosis of primary ciliary dyskinesia in consanguineous families. Sci Rep. 2017;7(1):7905.
Gao X, Su Y, Guan LP, Yuan YY, Huang SS, Lu Y, Wang GJ, Han MY, Yu F, Song YS, et al. Novel compound heterozygous TMC1 mutations associated with autosomal recessive hearing loss in a Chinese family. PLoS ONE. 2013;8(5):e63026.
Andersen TA, Troelsen KL, Larsen LA. Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci. 2014;71(8):1327–52.
Yang Y, Zhang W, Wu Q, Gao L, Jin W, Zhao T. Transcatheter closure of atrial septal defects without fluoroscopy: a well-established procedure for alternative use in children. Eurointervention. 2016;12(5):e652–7.
Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. 2017;120(6):908–22.
Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112(4):707–20.
Pawlak M, Niescierowicz K, Winata CL. Decoding the heart through next generation sequencing approaches. Genes (Basel). 2018;9(6):289.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–11.
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–5.
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–55.
Patient RK, McGhee JD. The GATA family (vertebrates and invertebrates). Curr Opin Genet Dev. 2002;12(4):416–22.
Chen J, Qi B, Zhao J, Liu W, Duan R, Zhang M. A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene. 2016;575(2 Pt 2):473–7.
Yu Y, Lei W, Yang J, Wei YC, Zhao ZL, Zhao ZA, Hu S. Functional mutant GATA4 identification and potential application in preimplantation diagnosis of congenital heart diseases. Gene. 2018;641:349–54.
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M, et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A. 2005;135(1):47–52.
Han H, Chen Y, Liu G, Han Z, Zhao Z, Tang Y. GATA4 transgenic mice as an in vivo model of congenital heart disease. Int J Mol Med. 2015;35(6):1545–53.
Morrisey EE, Ip HS, Tang Z, Parmacek MS. GATA-4 activates transcription via two novel domains that are conserved within the GATA-4/5/6 subfamily. J Biol Chem. 1997;272(13):8515–24.
Williams MR, Perry JC. Arrhythmias and conduction disorders associated with atrial septal defects. J Thorac Dis. 2018;10(Suppl 24):S2940–4.
Ellesoe SG, Johansen MM, Bjerre JV, Hjortdal VE, Brunak S, Larsen LA. Familial atrial septal defect and sudden cardiac death: identification of a Novel NKX2-5 mutation and a review of the literature. Congenit Heart Dis. 2016;11(3):283–90.
Mattapally S, Singh M, Murthy KS, Asthana S, Banerjee SK. Computational modeling suggests impaired interactions between NKX2.5 and GATA4 in individuals carrying a novel pathogenic D16N NKX2.5 mutation. Oncotarget. 2018;9(17):13713–32.
Dupays L, Shang C, Wilson R, Kotecha S, Wood S, Towers N, Mohun T. Sequential binding of MEIS1 and NKX2-5 on the Popdc2 gene: a mechanism for spatiotemporal regulation of enhancers during cardiogenesis. Cell Rep. 2015;13(1):183–95.
Stennard FA, Costa MW, Elliott DA, Rankin S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau BG, et al. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev Biol. 2003;262(2):206–24.
Sepulveda JL, Vlahopoulos S, Iyer D, Belaguli N, Schwartz RJ. Combinatorial expression of GATA4, Nkx2-5, and serum response factor directs early cardiac gene activity. J Biol Chem. 2002;277(28):25775–82.
Acknowledgements
We thank all patients and their parents for participating in this study. Thanks for the China Scholarship Council (award to Shijun Hu for 1 year’s study abroad at the Munich heart center and award to Haisong Bu for 2 year’s study abroad at the Mayo Clinic).
Funding
This study was supported by the National Natural Science Foundation of China (81800290), Hunan Provincial Natural Science Foundation of China (2019JJ40441) and Hunan Provincial Innovation Foundation for Postgraduate (CX20190156).
Author information
Authors and Affiliations
Contributions
HB and SH conceived and designed the study and drafted the manuscript. HB, GS and YZ collected the data. YY and ZT were involved in data cleaning and verification. HB, TZ and SH analyzed the data and critically revised manuscript. All authors were involved in the final draft of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all family member participants, and the patient’s parents in the case of participants under the age of 16 to the use of his/her all the related images and information for scientific purposes. The study was approved by the Committee on Clinical Trials and Applications at the Second Xiangya Hospital.
Consent for publication
Written informed consent was obtained from all family members and the patient/parents/legal guardians for publication of this research and any accompanying images and videos. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
The detailed information of whole-exome sequencing data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bu, H., Sun, G., Zhu, Y. et al. The M310T mutation in the GATA4 gene is a novel pathogenic target of the familial atrial septal defect. BMC Cardiovasc Disord 21, 12 (2021). https://doi.org/10.1186/s12872-020-01822-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-020-01822-5