
Abstract
Background
Weeds reduce wheat yields in dryland farming systems. Herbicides such as metribuzin are commonly used to control weeds. However, wheat has a narrow safety margin against metribuzin. Standing crops such as wheat with weeds in the same field can also be killed by the same dose of metribuzin. Therefore, it is important to identify metribuzin resistance genes and understand the resistance mechanism in wheat for sustainable crop production. A previous study identified a significant metribuzin resistance wheat QTL, Qsns.uwa.4 A.2, explaining 69% of the phenotypic variance for metribuzin resistance.
Results
Two NIL pairs with the most contrasting performance in the metribuzin treatment and different in genetic backgrounds were compared using RNA sequence analysis, identifying nine candidate genes underlying Qsns.uwa.4 A.2 responsible for metribuzin resistance. Quantitative RT-qPCR further validated the candidate genes, with TraesCS4A03G1099000 (nitrate excretion transporter), TraesCS4A03G1181300 (aspartyl protease), and TraesCS4A03G0741300 (glycine-rich proteins) identified as key factors for metribuzin resistance.
Conclusion
Identified markers and key candidate genes can be used for selecting metribuzin resistance in wheat.
Similar content being viewed by others
Background
Wheat is a major cereal crop worldwide and one of Australia’s most significant agricultural trade commodities. However, weed infestation costs Australian agriculture AUD 2.5–4.5 billion per annum [1], equivalent to ~ 20% of the gross value. Weeds are a major problem in Mediterranean agriculture [2, 3] and generally grow actively during the wet winter season. Weed control is essential to ensure crops can fully use stored summer rainfall and prevent weed seeds from contaminating crop seeds. Controlling weeds using broad-spectrum herbicides such as metribuzin, is common in agriculture [4]. Metribuzin—a triazine herbicide that targets PS-II components—is used primarily as a pre-emergent herbicide [4]. In Western Australia, pre-emergent application of Metribuzin controlled 80% of germinating weeds in demonstration trials [5]. However, other factors such as resistant sources (varieties), soil type, herbicide dose, field moisture, ambient temperature, and growing degree days [6] can determine the effectiveness of herbicides. Despite having limitations to use herbicides, herbicides are regularly used in controlling weeds. While using herbicide such as metribuzin for controlling weeds it can also kill germinating wheat from the same field. Therefore, it is essential to investigate wheat’s metribuzin resistance traits and underlying genes.
RNA sequencing of NILs can identify differentially expressed genes in wheat, as isolines of each NIL pair are essentially fixed, thus differences between isolines from a locus or few loci can be obtained accurately by analysing the NILs. Comparing isolines of different genetic backgrounds ensures the identification of genes common to different NIL pairs; as both NIL pairs are contrasting for the same locus. At the same time, it helps reduce background noises. Therefore, NIL analysis using targeted QTL could be the best for characterising metribuzin resistance.
QTL for metribuzin resistance have been mapped on chromosome 4 A in two wheat populations: Chuan Mai 25 (Resistant, R) × Ritchie (Susceptible, S) [7] and Synthetic W7984 × Opata M85 [8] populations. The genetic distance of the QTL is 49.6–58.3 cM between two flanking markers, Xbarc170 and Xbarc350, with a physical distance of 130.84 Mb according to the wheat reference genome [9]. The original QTL region contains ten SSR markers, apart from the flanking markers [8]. However, QTL markers may not be suitable for direct gene characterisation, pyramiding and marker-assisted programs [10]; developing NILs using a heterogeneous inbred family method can ensure NIL pairs with different genetic backgrounds, as each pair is a descendent progeny from an F2 individual [11] such contrasting NIL pairs in comparative analysis can help to establish accurate marker-trait association. Several studies have used this method to develop NILs and characterise important major QTL through transcriptome profiling, thus refining the mapping resolution of the targeted QTL [12,13,14] for increasing selection accuracy. This study investigated differentially expressed genes (DEGs) and molecular markers for metribuzin resistance from two NIL pairs from different genetic backgrounds, allowing us to detect the most significant candidate genes and underlying marker-trait associations.
Our previous study identified an SSR marker (Xbarc343) from Qsns.uwa.4 A.2 in a Synthetic W7984 ⋅ Opata M85 population, which was used to develop NILs in a Chuan Mai 25 ⋅ Ritchie population. Phenotypic characterisation confirmed that the resistance (R) allele from R-isolines increased metribuzin resistance by 63–85% (average 69%) compared with the susceptibility (S) allele from S-isolines. Thousand-grain weight co-segregated with metribuzin resistance, signifying the importance of this locus and its underlying gene(s) [15]. Similarly, NILs (NIL_3 and NIL_17) with different genetic backgrounds were used in RNA sequencing and real-time quantitative polymerase chain reaction (RT-qPCR) between isolines to identify key candidate genes and molecular markers for metribuzin resistance. This study aimed to (1) identify DEGs as candidate genes underlying Qsns.uwa.4 A.2, (2) explore the mechanism of metribuzin resistance by analysing the networks of the candidate genes, and (3) identify molecular variants and candidate genes for metribuzin resistance selection in wheat.
Results
Differentially expressed genes (DEGs) common between two NIL pairs
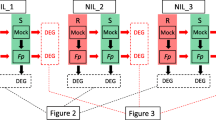
Within the control group, 199 genes were detected across both NIL pairs, while 297 genes were detected across the NILs within the treatment group, including DEGs (significant expression at P≤0.05) and non-DEGs (differences in gene expression between isolines of pairs were found but found not significant in differential expression) (Table 1). Twelve DEGs were found common between NIL_ 3 and NIL_17 on chromosome 4 A for metribuzin resistance (Fig. 1). Common DEGs in the two NIL pairs from the targeted 4 A QTL were used for metribuzin resistance analysis. Genes TraesCS4A03G0812600, TraesCS4A03G0819500, and TraesCS4A03G0912300LC were consistently downregulated (S-isolines had higher expression than R-isolines) in the metribuzin treatment but not detected as DEGs in the control. Three significantly different upregulated genes (R-isolines had higher expression than S-isolines) (P≤0.05) were located just outside the 4 A QTL (Table S1): TraesCS4A03G0542300, TraesCS4A03G0558000, and TraesCS4A03G0741300 (Table 2). Similarly, TraesCS4A03G1099000, TraesCS4A03G1171000, and TraesCS4A03G1181300 were upregulated (R-isolines had higher expression than S-isolines) and significantly differed between isolines in the metribuzin treatment within the 4 A QTL. These nine DEGs identified within and near the 4AQTL in NIL pairs can be considered as candidate genes for metribuzin resistance. The details of their molecular functions are in Table 2 and Table S1. However, the control and treatment groups had no common DEGs within the targeted QTL, as treatment DEGs did not significantly differ (P > 0.05) from control DEGs.

Twelve common DEGs were detected on chromosome 4 A between NIL_3 and NIL_17 as putative candidate genes responsible for metribuzin tolerance in wheat
In the control, three DEGs—TraesCS4A03G0781400 (TraesCS4A02G312800), TraesCS4A03G0990600 (TraesCS4A02G398300), and TraesCS4A03G1005600 (TraesCS4A02G404400)—were detected in both NIL pairs and significantly downregulated in the control, but not detected in either of the NIL pairs after the metribuzin treatment. This could be due to higher gene expression in R-isolines in response to treatment, resulting in non-DEGs (differ in gene expression but not-significant, P > 0.05) between the R-isolines and S-isolines. This outcome strongly suggests that differences in genetic backgrounds between isolines may cause different gene expressions in response to metribuzin treatment.
Single nucleotide polymorphisms (SNPs) between R and S isolines
We detected 4,051 SNPs in the R-isolines and S-isolines from all chromosomes. Five SNPs occurred within the 4 A QTL (Table 3; Tables S2 and S3) that mostly differed between the R-isolines and S-isolines, with three detected within genes (non-DEGs) and two found close to the candidate genes.
A major SNP common between two NIL pairs occurred near (0.10 Mb away) the candidate gene TraesCS4A03G1099000. Another SNP was found 0.015 Mb from candidate gene TraesCS4A03G1181300 (Table 3; Tables S2 and S3) but was not polymorphic between all R-isolines and S-isolines and thus cannot be considered a true SNP (Table 3). No other variants from 4 A chromosome common to NIL pairs were detected (Table 3; Tables S1–S3).
Gene ontology and functional annotation of DEGs
Biological processes, cellular components, and molecular functions were used to describe GO functions. The highly expressed genes in the metribuzin treatment are presented with their molecular functions and biological processes (Fig. 2). The most prevalent processes were biological processes, localisation, metabolic processes, response to stimuli, signaling and cellular processes. The most common cellular components were extracellular regions, macromolecular complex, membranes, and organelle parts. The most common molecular functions—binding, catalytic and transporter activities (Fig. 2 and Table S4)—were found to be significant in the metribuzin treatment.

Gene function classification (GO) based on biological function, cellular components, and molecular function. The most relevant biological processes in common DEGs are responses to stimulus, localisation, and signaling. The most relevant to cellular components are extracellular region and macromolecular complexes, while those most relevant to molecular functions are specific site binding and catalytic and transporter activities (indicated with arrows)
For the metribuzin treatment, KEGG pathway enrichment analysis of DEGs in isolines identified common pathways for glyoxylate and decarboxylate metabolism(KEGG ID; bdi00630), and nitrogen metabolism(KEGG ID; bdi01200) (Fig. 3). The phenylpropanoid and mitogen-activated protein kinase (MAPK) pathways(KEGG ID; bdi00940 and bdi04016, respectively) were the most significantly stimulated pathways for NIL_3. In contrast, alpha-linolenic acid and nitrogen metabolism were the most stimulated pathways for NIL_17 (Fig. 3).

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes (DEGs) in near-isogenic lines in the control and metribuzin treatments. (a) NIL_3 in the control; (b) NIL_3 in the metribuzin treatment; (c) NIL_17 in the control; (d) NIL_17 in the metribuzin treatment. Padj. is the corrected probability (P) value; red dots indicate highly significant enriched pathways (Padj.≤0.01), light yellow dots indicate significantally enriched pathways (Padj.≤0.05). Arrows indicate the pathways common to NIL_3 (b) and NIL_17 (d) pairs; KEGG analysis significantly enriched nitrogen metabolism and glyoxylate-decarboxylate metabolism in the metribuzin treatment [67]
Candidate gene validation using RT-qPCR
Nine candidate genes that significantly differed between isolines in the RNA sequencing-based DEG analysis from 4 A QTL (Table 2) were used in RT-qPCR-based DEG analysis (Fig. 4; Table S1 and Table S5). In the RT-qPCR analysis, DEGs were categorised based on the ratio of relative quantification (RQ) in gene expression between R-isoline and S-isoline significance at P≤0.05 (Table S5) using the paired t-test. Among the nine candidate genes, TraesCS4A03G1099000 (nitrate excretion transporter) was upregulated significantly, whereas TraesCS4A03G1181300 (aspartyl protease) and TraesCS4A03G0741300 (glycine-rich proteins) were upregulated (but non-significant) in both NIL pairs.

Gene expression analysis using RT-qPCR in NIL_3 and NIL_17 in the form of relative quantification (RQ) using dotplots (blue dots). 3R_T and 3S_T are the RQ of gene expression in resistant and susceptible isolines of NIL_3 (treatment), 17R_T and 17S_T are the RQ of gene expression in resistance and susceptible isolines of NIL_17 (treatment), 3R_C and 3S_C are the RQ of gene expression in resistance and susceptible isolines of NIL_3 (control), and 17R_C and 17S_C are the RQ of gene expression in resistance and susceptible isolines of NIL_17 (control). G1, G2, G3, G4, G5, G6, G7, G8, and G9 are the genes used for gene validation using RT-qPCR: TraesCS4A03G0812600 (G1), TraesCS4A03G0819500 (G2), TraesCS4A03G0912300LC(G3), TraesCS4A03G1099000 (G4), TraesCS4A03G1171000 (G5), TraesCS4A03G1181300 (G6), TraesCS4A03G0542300 (G7), TraesCS4A03G0558000 (G8), and TraesCS4A03G0741300 (G9)
Gene network analysis using candidate genes
In the control, the major DEGs—TraesCS4A03G0781400, TraesCS4A03G0990600, and TraesCS4A03G1005600 (named TraesCS4A02G312800, TraesCS4A02G398300, and TraesCS4A02G404400 in RefSeq1.1, respectively)—were used for network analysis. In the metribuzin treatment, the major DEGs—TraesCS4A03G1099000, TraesCS4A03G1181300, TraesCS4A03G0558000, and TraesCS4A03G0741300 (named TraesCS4A02G440700, TraesCS4A02G469600, TraesCS4A02G210100, and TraesCS4A02290900 in RefSeq1.1, respectively)—showed consistent expression patterns in the RNA-seq and RT-qPCR experiments and were used for network analysis. The gene network analysis revealed the interaction of these genes for stress resistance, including disease resistance, and their contribution to traits such as seed size, root morphological traits, and protein content (Fig. 5).

Gene network analysis using DEGs obtained from the control (A) experiment: TraesCS4A02G312800, TraesCS4A02G398300, and TraesCS4A02G404400, and DEGs obtained from the treatment (B) experiment: TraesCS4A02G440700, TraesCS4A02G469600, TraesCS4A02G210100, and TraesCS4A02290900. Triangle, genes directly associated with metribuzin resistance; hexagon, gene isoforms; square, phenotypes may relate to metribuzin resistance; red circle, proteins; dark green star, interaction network of resistance genes; red lines, protein–protein interactions that may interact with metribuzin resistance; light red lines, interaction of phenotypes may relate to metribuzin resistance
Discussion
Differentially expressed candidate genes underlying the Qsns.uwa.4 A.2 region
Using two NIL pairs with different genetic backgrounds in the DEG analysis using RNA-seq, we identified nine candidate genes and SNPs from Qsns.uwa.4 A.2 which identified six upregulated (higher gene expression in R-isolines than S-isolines) and three downregulated (higher expression in S-isolines than R-isolines) candidate genes near and within the Qsns.uwa.4 A.2, which were validated by RT-qPCR. The candidate genes are related to carbon metabolism, lipid metabolism, protein metabolism, and stress resistance. No common DEGs between the treatment and control were detected on chromosome 4 A (Table S1), suggesting that the applied metribuzin affected isolines gene expression. The DEGs found common between two NIL pairs from all wheat chromosomes are listed in Table S6.
Recent studies using Synthetic W7984 × Opata M85 QTL mapping [8], transcriptomic analyses of Chuan Mai 25 and Ritchie parents [16], and QTL mapping by DArT sequencing and genotyping in Chuan Mai 25 × Ritchie population [7] reported candidate genes for metribuzin resistance in wheat, mainly associated with photosynthesis, photosystem, and transporters related to the biosynthesis of fructose, mannose, galactose, and nucleotides. Similar functional categories genes are discussed in the following paragraphs.
Candidate gene TraesCS4A03G0812600 is an ethylene-responsive factor (ERF) with domain (AP2/ERF). The ERF domain has diverse roles in biotic and abiotic stresses [17]. TraesCS4A03G0812600 has the conserved domain amino acid sequences (rgvrkrpwg) at the 14th position, possibly the gene is ERF-1. ERF-1 expression depends on S-adenosyl- methionine (SAM) whose regulation is usually determined by the metabolism of methionine [18]. The major roles of SAM are regulating DNA methylation and 1-aminocyclopropane-1-carboxylic acid (ACC) synthase. In the absence of the ACC molecule, inactivated ETR-1 regulates the degradation of the C-terminal end of EIN-2 (a protein kinase) resulting in constitutive triple response 1 (CTR1) expression, helping in stopping ethylene production [19, 20]; whereas higher expression of ctr1 gene prevents EIN-3 degradation and promotes further ethylene signaling and biosynthesis, and thus may enhance leaf senescence. Dihydroxymethylchlorophyllide is a methyl derivative, is synthesised from ethylene, actively involves in chlorophyll biosynthesis, plays a significant role in chlorophyll subunit (CHLH) of Mg-chelatases [21] and seperation, and eventually degrading the green pigments from leaves. However, gene expression result was found inconclusive from RT-qPCR for this gene as there was no significant differences between isolines (Table S5 and Fig. 4). From RNA-seq, gene expression was found significantly different in isolines and found common to the isolines of both NIL pairs, we speculated that this gene may express significantly different between isolines when severe and longer metribuzin stress is imposed, when degradation of chlorophyll is started.
The gene TraesCS4A03G0819500 had higher expression in S-isolines than R-isolines with molecular functions similar to leucine-rich repeat (LRR)-serine/threonine-protein kinase. This gene may recognise and transduce metribuzin signals toward the effectors. LRR regulates various abiotic and biotic stresses in plants [22,23,24], with a major role in transducing metribuzin signals, through the membrane, triggering the MAPK cascade for synthesising jasmonic acid, flavonoids, and phenylpropanoids [25, 26] and determining ethylene signaling. Ethylene response factor, a downregulated candidate gene (Table 2), also regulates the constitutive triple response-1 (ctr1), which has a functional domain similar to serine/threonine protein kinase [26, 27], and may closely resemble kinase activities, such as phosphorylation in serine and threonine (ST) residues [27, 28]. This process of phosphorylation leads to conformational changes [29, 30]. A gene ST8 on PSII-component phosphorylation/activation-deactivation has been reported [31]. When the metribuzin signal passes through phosphorylation, mutation of the Psb gene and substitution of amino acids may occur at the Ser-264-Thr site [32], which codes the D1 protein, affecting the efficiency of electron transport and ATP synthesis. ATPs are important for cell differentiation, photosynthetic processes, cell growth, and various cellular metabolism.
Candidate gene TraesCS4A03G1171000 is a resistance gene analog-5 (RGA-5), expressed more in R-isolines than S-isolines. Plant resistance gene analogs (RGAs) also have diverse roles in determining biotic and abiotic resistance in plants [33]. Well-known RGAs are nucleotide-binding site leucine-rich repeats (NB-LRR), receptor-like kinases, and receptor-like proteins. These protein-encoding genes are responsible for receiving signals through receptors and transducing signals to nucleotide-binding site(s) for transcription [34] [35]. Similarly, the upregulated NB-LRR gene also produces ROS, involved in lignin and callus biosynthesis [36]. The lignification-related gene TraesCS4A03G0558000 was also upregulated in both NIL pairs but detected outside the QTL (Table S1). These complex biopolymers (lignin and calluses) may be used as energy suppliers through the tricarboxylic acid cycle (TCA) when plants are under stress and may also help with the turnover of PS-II components to their original state. Succinyl CoA, an intermediate of TCA cycle with amino acid glycine initiate the synthesis of chlorophyll-A [37] during chlorophyll biosynthesis, therefore, TraesCS4A03G0558000 may also help in chlorophyll biosynthesis.
An annotation of the common (between two NIL pairs) downregulated TraesCS4A03G0912300LC was unavailable. The sequence similarity analysis, GO, UniProt search, NCBI blast, and knetminer analyses did not reveal any links to other genes. The candidate gene is present within 4AQTL with other candidate genes, indicating that they may follow a similar pathway of metribuzin resistance [38]. Therefore, we assume the gene is related to metribuzin resistance.
Candidate gene TraesCS4A03G1099000 showed upregulation with gene function related to the nitrate excretion transporter. The RT-qPCR and RNA-seq based DEG analysis revealed that this gene significantly differed between R-isolines and S-isolines in NIL_3 (Table S5, Fig. 4, and Table 2). None of the other candidate genes significantly differed in their expression between isolines; therefore, this gene may be the first responsive gene to metribuzin (e.g., signal-perceiving or signal-activating). The functional role of this gene in plants is directly associated with stress response, signaling, growth, and metabolism of peptides and proteins [39]. Carbohydrate and protein metabolisms are also related to determining the nitrate level in plants. Photosynthate portioning, carbon–nitrogen balance, maintaining cell homeostasis, signal transduction, and nucleotide synthesis and repairs are crucial processes under stress conditions [40, 41] and related to nitrogen metabolism. Similarly, functional roles of anions with the oxygen-evolving complex, a PS-II component, have been reported; the efficiency of anions with CaMn4O5 of PS-II were ordered Cl > Br > NO3 > NO2 > I [42,43,44]. It has been reported that NO2 is oxidised by the OEC of PS-II, thus inhibits the PS-II [45]. Hebicides including of photosynthestic inhibitors also interfere in the normal nitrate reduction process which can result in nitrate accumulation in PS-II [46], which may also hamper the ammonia assimilation in the cells. Important gene/protein such as Qb site from PS-II, a reported site for herbicide binding [47], could be the same site where nitrate may interact for metribuzin resistance. The overly produced nitrates in plant cells under metribuzin stress may change the expression of gene TraesCS4A03G1099000 (nitrate excretion transporter), may help to release nitrate from PS-II for ammonia assimilation process [48, 49] and signaling other linked genes such as nitrite reductases (NR), glutamine synthetase (GS), glutamate synthase (GOGAT) [50], and chlorophyll biosynthesis [51,52,53,54,55]. An SNP (marked bold in Table 3) identified near the candidate gene TraesCS4A03G1099000 could be used to select for metribuzin resistance in wheat. While this SNP was not polymorphic between R-isolines and S-isolines of NIL_17 under metribuzin treatment (Table S2)—possibly because the metribuzin effects at this SNP site result in the substitution of DNA sequences from G to A in isoline 17S_T—this change may contribute to metribuzin resistance in isoline 17S_T. This mutation could be one of the reasons explaining why TraesCS4A03G1099000 expression did not significantly differ in contrasting isolines of pair NIL_17 in the RT-qPCR. However, this SNP was polymorphic between R-isolines and S-isolines in NIL_3; mutation (A to G) at this SNP site in NIL_3 (Table S3 and Table 3) may favour to bind with metribuzin in 3S_T isoline, with significant differences in gene expression (TraesCS4A03G1099000) observed between isolines in NIL_3. Gene TraesCS4A03G1099000 was also closely associated (Table 3) with marker, Xbarc343 (physical position 694,903,704 bp on 4 A chromosome), which was used in NIL development [15] for metribuzin resistance, and SNP site was detected (in this current research) at 714,355,066 bp; therefore, the marker distance of 19.45 Mb has decreased from 694,903,704 bp (original Xbarc343 position) to 714,355,066 bp (current SNP position). This SNP position at 714,355,066 bp is 0.10 Mbp away from the candidate gene TraesCS4A03G1099000, may affect the metribuzin resistance. Using this SNP as a molecular marker, other key linked variants can be identified using higher resolution mapping for selecting metribuzin resistance.
Upregulated candidate gene TraesCS4A03G1181300 was common to both NIL pairs, with the most significant differences in gene upregulation from RNA-seq (Table 2) and upregulated through RT-qPCR analysis (Table S5). It has a function related to aspartyl protease [56] found in chloroplast membranes and stroma [57], and its processing is senescence-related [58]. Aspartate, a amino acid closely resembles to glutamate, are frequently transaminated [59] during amino acid synthesis, while glutamate is a precurser for chlorophyll synthesis [60].
Another candidate gene TraesCS4A03G0741300 was responsive to stress; the functional annotation revealed that the gene encodes glycine-rich protein, related to stress resistance and protein processing [60, 61]. During chlorophyll biosynthesis succinyl CoA, an intermediate of Krebs cycle and the amino acid glycine initiate the synthesis of chlorophyll-A leading to production of protochlorophyllide or protochlorophyll [62,63,64] which are the initiating molecules in chlorophyll biosynthesis. This gene was significantly upregulated in the RNA-seq and upregulated in RT-qPCR in both NIL pairs (G9 gene in Fig. 4), suggesting an important role in chlorophyll synthesis and metribuzin resistance in wheat.
Another upregulated DEG common between two NIL pairs is TraesCS4A03G0558000 with annotation function is related to divalent metal-binding) (Table S1). Gene TraesCS4A03G0558000 could be the Mg++ion of chlorophyll porphyrin ring, because it likely follow the same pathway of chlorophyll synthesis and metribuzin resistance as with other candidate genes from the selected QTL. Mg++ binding protein (gene TraesCS4A03G0558000) makes bonds with methyl groups and four nitrogen atoms in square planner arrangement in chlorophyll molecule [65]. Gene higher expression may directly be associated with the formation of chemical energy through the process of [66]. However, gene was not found commonly expressed to both pairs in RT-qPCR (G8 gene in Fig. 4), due to differential level of chlorophyll may present in NIL pairs leaves after 15 days of metribuzin treatment.
Gene network analysis using DEGs from the control and metribuzin treatments revealed that candidate genes were responsive to stresses such as disease and some agronomically important traits such as grain size, root traits, and protein content (Fig. 5). Our previous study [15] found that the 4 A QTL responsible for metribuzin resistance was closely linked to thousand-grain weight. Similarly, the network analysis in the present study also found that the key candidate genes underlying 4AQTL may play roles in regulating grain size, suggesting that targeting this QTL may improve metribuzin resistance and the yield component trait simultaneously.
Candidate genes responsive to metribuzin treatment, genes such as TraesCS4A03G1099000 (nitrate excretion transporter), may act as a precurser for chlorophyll synthesis genes, may help to activate the genes TraesCS4A03G1181300 (aspartyl protease), TraesCS4A03G0558000 (divalent metal binding protein), TraesCS4A03G0741300 (glycine-rich protein) and TraesCS4A03G0812600 (ethylene-responsive). During metribuzin stresss, gene TraesCS4A03G1099000 may get express and release from PS-II in response to stress, and may activate the other precursers such as aspartate, glycine, metal binding protein, and ethylene responsive genes for chlorophyll synthesis which eventually may help in photosynthetic carbon assimilation and metribuzin resistance which is also revealed from the thousand grain weight (TGW) and metribuzin resistance were found closely linked genes in [15]. Therefore, it can be said while selecting for the gene TraesCS4A03G1099000, using linked molecular marker, we can able to balance the carbon-nitrogen content in wheat, which ultimately helps in carbon assimilation, chlorophyll enhancement and metribuzin resistance in cyclic manner. Other candidate genes such as TraesCS4A03G0819500 (leucine-rich repeat (LRR)-serine/threonine-protein kinase) and TraesCS4A03G1171000 (nucleotide-binding (NB)-LRR receptor kinase), may also be involved in pathways of metribuzin resistance when longer duration of metribuzin dose is applied.
The KEGG analysis [67,68,69] reveaed that the candidate genes were involved in nitrogen metabolism and carbohydrate metabolism (Fig. 3) which were accordance with the results (candidate genes function; Table 2) obtained in DEG analysis, indicating that candidate genes are involved in carbon-nitrogen related pathways for metribuzin resistance. Genes that significantly differed in their expression in NILs in the RNA-seq but not validated in the RT-qPCR analysis may still be involved in metribuzin resistance pathways, requiring more metribuzin exposure time to express differentially between isolines. Therefore, further a time-point experiment using RT-qPCR is needed to validate other candidate genes for metribuzin resistance in wheat. This study reported and validated DEGs using RNA-seq and qPCR, which indicated that TraesCS4A03G1099000 (nitrate excretion transporter), TraesCS4A03G1181300 (aspartyl protease), and TraesCS4A03G0741300 (glycine-rich proteins) are the major genes for metribuzin resistance in wheat.
Variants in the two NIL pairs
Five variants within the 4 A QTL differed between the isolines for both NIL pairs, including three SNPs within the genes (non-DEGs) and two SNPs away from candidate genes. SNPs common between isolines of NIL pairs are the most valuable markers as they also indicate similar genomic composition between two different genetic backgrounds from the targeted genomic regions.
A major SNP located near gene TraesCS4A03G1099000 can be considered a marker, which was within gene TraesCS4A03G1098900 (Table 3). Both these genes code for the nitrate excretion transporter. This SNP overlapped in isolines of NIL pairs (Table S3), which could affect the expression of candidate gene TraesCS4A03G1099000 and may also be linked with other candidate genes. The SNP near the candidate gene (< 1Mbp) or candidate gene regulatory regions could be valuable markers for selecting metribuzin resistance. The alleles can be used for fine mapping and gene validation experiments for metribuzin resistance in wheat.
Conclusion
Transcriptomic analysis using NILs targeting major 4AL QTL Qsns.uwa.4 A.2—explaining up to 69% of the phenotypic variance for metribuzin resistance in wheat—revealed nine candidate genes and five consistent SNPs associated within the QTL. Among the candidate genes; TraesCS4A03G1099000 (nitrate excretion transporter) and TraesCS4A03G1181300 (aspartyl protease) and TraesCS4A03G0741300 (glycine-rich protein) are likely major candidates for determining metribuzin resistance based on the consistent RNA-seq and RT-qPCR results. The detected SNP markers at 714,355,066 bp on wheat chromosome 4 A might be used to select important candidate gene TraesCS4A03G1099000 for metribuzin resistance which may also ultimately help in yield enhancement in wheat.
Methods
Plant materials and metribuzin treatment
Two NIL pairs with different genetic backgrounds—3R and 3 S and 17R and 17 S—were used in this study (R-isolines are resistant isolines with the R allele, and S-isolines are susceptible isolines with the S allele) [15]. NILs were developed from a cross of Chuan Mai 25 and Ritchie. Chuan Mai 25 is a Chinese cultivar derived from the cross of 1414/Chuanyu 5//Genaro 80 (https://repository.cimmyt.org/xmlui/bitstream/handle/10883/1222/64613.pdf, accessed on 10 October 2022), while Ritchie is a European cultivar released in the United Kingdom. The NILs were grown in three biological replicates in the glasshouse at The University of Western Australia (UWA), Perth, Western Australia. Seeds of each isoline were sown and sprayed with 200 g ai./ha pre-emergent dose of metribuzin (recommended dose to control weeds in Western Australia wheat fields). NILs seeds were kept in the Gene Bank, UWA, Perth. The plant phenotyping/metribuzin assessment method is described in [15]. Fifteen-day-old seedlings and roots were sampled for RNA extraction. NIL pair seeds were sown under 100% field capacity in river sand (no dirt or clay). Wheat seedlings (including roots) were used for the total RNA sample collection; for example, three seedlings of NIL_3_R/S and NIL_17_R/S were bulked for Replication-I, three seedlings of NIL_3_R/S and NIL_17_R/S were bulked for Replication-II, and three seedlings of NIL_3_R/S and NIL_17_R/S were bulked for Replication-III. The samples were placed in chilled 50 mL autoclaved tubes, immediately capped, and immersed into a liquid nitrogen dewar at − 197° C to extract samples total RNAs from NIL pairs—3R_T, 3S_T, 3R_C, 3S_C, and 17R_T, 17S_T, 17R_C, and 17S_C—for gene expression analysis.
RNA extraction, library preparation, and sequencing
Total RNA was extracted from 24 samples (four isolines × three replicates × two treatments) using an RNeasy Plus Plant Mini Kit (Qiagen) with DNase-I treatment, following the manufacturer’s instructions (Bioline, Australia). The quantity and purity of extracted RNA were assessed by NanoDrop 2000 (Thermo Fisher Scientific Inc., Australia). The cDNA synthesis, 150 bp paired-end sequencing, raw data filtering, and other sequencing parameters were set as the default, as per the Novogene protocol (en.novogene.com). Clean data were generated as FastQ files, with clean read quality checking (Q20 and Q30) and GC contents quantified. Clean sequencing data from NIL pairs were used for gene expression and DEG analysis. Paired-end sequencing data from all samples were deposited to the National Center for Biotechnology Information (NCBI) in SRA, with an accession number of PRJNA839200.
Transcriptome assembly and mapping quality statistics
The Illumina NovaSeq6000 platform generated 183 Gb high-quality 150 bp pair-end sequencing reads from the 24 samples after quality control, with an average of 76 million clean reads per library. The percentage of bases with Phred quality scores of Q20 and Q30 were 98% and 94%, respectively (Table S7). Approximately 86% of the sequenced reads were mapped to the wheat reference genome, of which 89% per library uniquely matched. The GC base percentage was 54% of the total bases. The sequencing error rate was < 0.02, calculated using the Qphred parameter as the default, as per the Novogene protocol. The fragments per kilobase million (FPKM) mapped reads detected in each library ranged from 98,253 to 117,430 (average 111,032), accounting for 80% of wheat genes. Pearson’s correlation coefficients for each combination of samples within biological samples ranged from 0.8 to 0.9, indicating there was a minimal variation between biological replicates.
Sequencing, analysis, and differentially expressed gene (DEG) identification
The obtained sequences were aligned to the reference sequences (RefSeq V2.1) using the HISAT2 algorithm [70, 71]. Bowtie2 was used to align the reads to the Chinese Spring wheat reference transcriptome [72], with the gene expression level calculated using RSEM (RNA-Seq by Expectation Maximisation) software v1.2.1246 with default parameter [73]. The expected number of transcripts was reported in FPKM. DEGs were detected with DEGseq as described in Wang et al. [62] with the following parameters: differential genes are Log2fold change > 1 for upregulated genes, and Log2fold change <–1 for downregulated genes at P≤0.05. Common candidate genes obtained from different NIL pairs are those in which DEGs from isolines show similar gene expression patterns (upregulation or downregulation) from both pairs. The molecular functions of DEGs were also validated with the NCBI nucleotide search by blasting the gene sequences.
Gene ontology and functional pathways analysis
Gene ontology (GO) enrichment analysis of DEGs was implemented using the clusterProfiler v4.0 R package with bias correction [74]. Significantly enriched GO terms had corrected P ≤0.05. The Kyoto Encyclopedia of Genes and Genomes (KEGG)[67,68,69] was used for studying pathway enrichment to understand more functions (https://www.genome.jp/kegg).
Variant (SNPs and indels) identification
The reference sequence from IWGSC_RefSeq_v2.1 was used to align the sample sequences for identifying variants [71] (SNPs and Indels) (Table S2 and Table S3). Variant calling was performed using GATK software v4 [75], and SnpEff software v5 was used to annotate the variant sites [76]. Specifically, after applying GATK to detect variant sites, each site was counted according to the SnpEff annotation information; for example, the variant site function was plotted statistically from three aspects: non-synonymous mutation, missense mutation, and nonsense mutation. Variants were identified by comparing R-isoline or S-isoline sequences with the reference sequences; those common to each set (three replicates) of R-isolines or S-isolines and also differed between R-isolines and S-isolines were considered true variants.
DEGs validation using RT-qPCR analysis
Candidate genes in the targeted Qsns.uwa.4 A.2, commonly expressed (upregulated or downregulated) between the two NIL pairs, were selected for RT-qPCR assay. RNA concentrations of the samples were measured with NanoDrop1000 Spectrophotometer (ThermoScientific) and adjusted accordingly. Total RNA was extracted from 24 samples using a Qiagen RNeasy Plant Mini Kit according to the protocol followed by Md. Sultan Mia [77]. Total RNA (1 µg) was used to synthesise cDNA using a SensiFAST cDNA Synthesis Kit (Bioline Australia) following the manufacturer’s protocol. The RT-qPCR was performed on an ABI 7500 Fast system using a SensiFAST™ SYBR No-ROX Kit (Bioline Australia). Gene-specific primers were designed based on candidate gene exon sequences with the help of Geneious software, with the wheat actin gene [78] used as an endogenous control to normalise gene expression. Primers were designed from the concatenated sequence regions covering the exon–exon junctions to avoid residual gDNA amplification. The function of the designed primers was checked with NCBI nucleotide blast to ensure the selected primers represent the targeted genes (Table S5). Three biological replicates were used in each isoline from two NIL pairs for treatment and control conditions. Amplification was conducted in a 20 µL reaction mix containing 10 µL of 2×SensiFAST SYBR Lo-ROX mix, 0.8 µL of 10 µM each for forward and reverse primers, 100 ng cDNA (usually 1 µL), with the following cycling protocol: 1 cycle of 95° C for 2 min, 40 cycles of 95° C for 5 s, and 63° C for 30 s. Ct mean values were calculated based on differences between the endogenous control and each sample value (ΔΔCт) converted to Log2 values, representing the relative quantification (RQ) for gene expression [79]. The observed values of relative quantification were analysed using paired two sample t-test. Differences in gene relative quantification between R-isolines and S-isolines determines gene upregulation (positive values)and downregulation (negative values). The parameter was set at P ≤ 0.05 for the paired t-test.
Data Availability
The datasets generated and/or analysed (RNA-seq data) during the study are available in the NCBI repository with Bioproject number PRJNA839200 (released on 9 October 2022). Other data such as SNP information are available in the supplementary tables.
Abbreviations
- AP2/ERF:
-
AP2-like ethylene-responsive
- 4AL:
-
Long arm of chromosome 4 A
- ATP:
-
Adenosine triphosphate
- DNase I:
-
Deoxyribonuclease I
- cDNA:
-
Complementary DNA
- DEG:
-
Differentially expressed gene
- ETR:
-
Ethylene response
- EIN:
-
Ethylene insensitive
- FPKM:
-
Fragments per kilobase of exon per million mapped reads
- G × E:
-
Gene by environment interaction
- GO:
-
Gene ontology
- HIF:
-
Heterogeneous inbreed family
- Indel:
-
Insertion and deletion
- IWGSC:
-
International Wheat Genome Sequence Consortium
- Kbp:
-
Kilobase pair
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- Mbp:
-
Megabase pair
- MQTL:
-
Meta-QTL
- NBS-LRR:
-
Nucleotide-binding site leucine-rich repeat
- NCBI:
-
National Center for Biotechnology Information
- NIL:
-
Near-isogenic line
- Q30:
-
Quality score of 30
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- QTL:
-
Quantitative trait locus
- RGA:
-
Resistance gene analog
- RNA-seq:
-
RNA sequencing
- ROS:
-
Reactive oxygen species
- S:
-
Susceptible
- SNT:
-
Serine/threonine protein kinase
- SNP:
-
Single nucleotide polymorphism
- SRA:
-
Sequence read archive
- R:
-
Resistant
- SAM:
-
S-adenosyl- methionine
- TFs:
-
Transcription factors
- UTR:
-
Untranslated region
- ΔΔCT:
-
Delta delta C (T) method
References
Roberts J, Florentine S. Biology, distribution and control of the invasive species Ulex europaeus (Gorse): a global synthesis of current and future management challenges and research gaps. Weed Res. 2021;61(4):272–81.
Ryan J, De Pauw E, Gomez H, Mrabet R. Drylands of the Mediterranean zone: biophysical resources and cropping systems. Dryland Agric. 2006;23:577–624.
Rose TJ, Parvin S, Han E, Condon J, Flohr BM, Schefe C, et al. Prospects for summer cover crops in southern australian semi-arid cropping systems. Agric Syst. 2022;200:103415.
Kraehmer H, Laber B, Rosinger C, Schulz A. Herbicides as weed control agents: state of the art: I. weed control research and safener technology: the path to modern agriculture. Plant Physiol. 2014;166(3):1119–31.
Khalil Y. Interaction of pre-emergent herbicides and crop residues in western australian no-tillage systems. The University of Western Australia; 2018.
Yamaji K, Watanabe Y, Masuya H, Shigeto A, Yui H, Haruma T. Root fungal endophytes enhance heavy-metal stress tolerance of Clethra barbinervis growing naturally at mining sites via growth enhancement, promotion of nutrient uptake and decrease of heavy-metal concentration. PLoS ONE. 2016;11(12):e0169089.
Xu L, Liu H, Kilian A, Bhoite R, Liu G, Si P, et al. QTL mapping using a high-density genetic map to identify candidate genes associated with metribuzin tolerance in hexaploid wheat (Triticum aestivum L). Front Plant Sci. 2020;11:573439.
Bhoite R, Onyemaobi I, Si P, Siddique KHM, Yan G. Identification and validation of QTL and their associated genes for pre-emergent metribuzin tolerance in hexaploid wheat (Triticum aestivum L). BMC Genet. 2018;19(1):1–11.
International Wheat Genome, Sequencing C, Appels R, Eversole K, Stein N, Feuillet C, Keller B, et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361(6403):eaar7191.
Collard BCY, Mackill DJ. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philosophical Trans Royal Soc B: Biol Sci. 2008;363(1491):557–72.
Boyles Iii RE. Genetic study of grain yield and quality traits in sorghum. Clemson University; 2016.
Ma J, Stiller J, Zhao Q, Feng Q, Cavanagh C, Wang P, et al. Transcriptome and allele specificity associated with a 3BL locus for Fusarium crown rot resistance in bread wheat. PLoS ONE. 2014;9(11):e113309.
Wang X, Liu H, Siddique KHM, Yan G. Transcriptomic profiling of wheat near-isogenic lines reveals candidate genes on chromosome 3A for pre-harvest sprouting resistance. BMC Plant Biol. 2021;21(1):1–14.
Nouraei S, Mia MS, Liu H, Turner NC, Yan G. Transcriptome analyses of near isogenic lines reveal putative drought tolerance controlling genes in wheat. Frontiers in Plant Science. 2022;13.
Bhattarai R, Liu H, Siddique KHM, Yan G. Characterisation of a 4A QTL for Metribuzin Resistance in Wheat by developing Near-Isogenic Lines. Plants. 2021;10(9):1856.
Bhoite R, Si P, Siddique KHM, Yan G. Comparative transcriptome analyses for metribuzin tolerance provide insights into key genes and mechanisms restoring photosynthetic efficiency in bread wheat (Triticum aestivum L). Genomics. 2021;113(3):910–8.
Xie Z, Nolan TM, Jiang H, Yin Y. AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in Arabidopsis. Front Plant Sci. 2019;10:228.
Watanabe M, Chiba Y, Hirai MY. Metabolism and regulatory functions of O-acetylserine, S-adenosylmethionine, homocysteine, and serine in plant development and environmental responses. Front Plant Sci. 2021;12:643403.
Guo F-Q, Young J, Crawford NM. The nitrate transporter AtNRT1. 1 (CHL1) functions in stomatal opening and contributes to drought susceptibility in Arabidopsis. Plant Cell. 2003;15(1):107–17.
Tang J, Chu C. MicroRNAs in crop improvement: fine-tuners for complex traits. Nat Plants. 2017;3(7):1–11.
Tripathy BC, Pattanayak GK. Chlorophyll biosynthesis in higher plants. Photosynthesis: plastid biology, energy conversion and carbon assimilation. 2012:63–94.
Van der Does D, Boutrot F, Engelsdorf T, Rhodes J, McKenna JF, Vernhettes S, et al. The Arabidopsis leucine-rich repeat receptor kinase MIK2/LRR-KISS connects cell wall integrity sensing, root growth and response to abiotic and biotic stresses. PLoS Genet. 2017;13(6):e1006832.
Huang L, Raats D, Sela H, Klymiuk V, Lidzbarsky G, Feng L, et al. Evolution and adaptation of wild emmer wheat populations to biotic and abiotic stresses. Annu Rev Phytopathol. 2016;54(1):279–301.
Ravindran P, Yong SY, Mohanty B, Kumar PP. An LRR-only protein regulates abscisic acid-mediated abiotic stress responses during Arabidopsis seed germination. Plant Cell Rep. 2020;39(7):909–20.
War AR, Paulraj MG, Ahmad T, Buhroo AA, Hussain B, Ignacimuthu S, et al. Mechanisms of plant defense against insect herbivores. Plant Signal Behav. 2012;7(10):1306–20.
Clark KL, Larsen PB, Wang X, Chang C. Association of the Arabidopsis CTR1 Raf-like kinase with the ETR1 and ERS ethylene receptors. Proceedings of the National Academy of Sciences. 1998;95(9):5401-6.
Edelman AM, Blumenthal DK, Krebs EG. Protein serine/threonine kinases. Annu Rev Biochem. 1987;56(1):567–613.
Pesaresi P, Pribil M, Wunder T, Leister D. Dynamics of reversible protein phosphorylation in thylakoids of flowering plants: the roles of STN7, STN8 and TAP38. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2011;1807(8):887–96.
Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109(3):275–82.
Molle V, Kremer L. Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol Microbiol. 2010;75(5):1064–77.
Bonardi V, Pesaresi P, Becker T, Schleiff E, Wagner R, Pfannschmidt T, et al. Photosystem II core phosphorylation and photosynthetic acclimation require two different protein kinases. Nature. 2005;437(7062):1179–82.
Wildner GF, Heisterkamp U, Trebst A. Herbicide cross-resistance and mutations of the psbA gene in Chlamydomonas reinhardtii. Z für Naturforschung C. 1990;45(11–12):1142–50.
Cortaga CQ, Latina RA, Habunal RR, Lantican DV. Identification and characterization of genome-wide resistance gene analogs (RGAs) of durian (Durio zibethinus L). J Genetic Eng Biotechnol. 2022;20(1):1–11.
Wu J, Yan G, Duan Z, Wang Z, Kang C, Guo L, et al. Roles of the Brassica napus DELLA protein BnaA6. RGA, in modulating drought tolerance by interacting with the ABA signaling component BnaA10. ABF2. Front Plant Sci. 2020;11:577.
Shi H, Liu W, Wei Y, Ye T. Integration of auxin/indole-3-acetic acid 17 and RGA-LIKE3 confers salt stress resistance through stabilization by nitric oxide in Arabidopsis. J Exp Bot. 2017;68(5):1239–49.
Balint-Kurti P. The plant hypersensitive response: concepts, control and consequences. Mol Plant Pathol. 2019;20(8):1163–78.
Broddrick JT, Rubin BE, Welkie DG, Du N, Mih N, Diamond S et al. Unique attributes of cyanobacterial metabolism revealed by improved genome-scale metabolic modeling and essential gene analysis. Proceedings of the National Academy of Sciences. 2016;113(51):E8344-E53.
Broekgaarden C, Snoeren TAL, Dicke M, Vosman B. Exploiting natural variation to identify insect-resistance genes. Plant Biotechnol J. 2011;9(8):819–25.
Flynn KJ. Algal carbon–nitrogen metabolism: a biochemical basis for modelling the interactions between nitrate and ammonium uptake. J Plankton Res. 1991;13(2):373–87.
Zhang Y, Zhou J, Yang Y, Elgamal WH, Xu P, Li J, et al. Two SNP mutations turned off seed shattering in Rice. Plants. 2019;8(11):475.
Tao D, Hu F, Yang J, Yang G, Yang Y, Xu P, et al. Cytoplasm and cytoplasm-nucleus interactions affect agronomic traits in japonica rice. Euphytica. 2004;135(1):129–34.
Kelley PM, Izawa S. The role of chloride ion in Photosystem II I. Effects of chloride ion on Photosystem II electron transport and on hydroxylamine inhibition. Biochim et Biophys Acta (BBA)-Bioenergetics. 1978;502(2):198–210.
Wydrzynski TJ, Satoh K, Freeman JA. Photosystem II: the light-driven water: plastoquinone oxidoreductase. Springer; 2005.
Kawakami K, Umena Y, Kamiya N, Shen J-R. Location of chloride and its possible functions in oxygen-evolving photosystem II revealed by X-ray crystallography. Proceedings of the National Academy of Sciences. 2009;106(21):8567-72.
Pokhrel R, Brudvig GW. Investigation of the inhibitory effect of nitrite on Photosystem II. Biochemistry. 2013;52(21):3781–9.
Klepper L. A mode of action of herbicides: Inhibition of the normal process of nitrite reduction. 1974.
Melis A. Photosystem-II damage and repair cycle in chloroplasts: what modulates the rate of photodamage in vivo? Trends Plant Sci. 1999;4(4):130–5.
Foyer CH, Noctor G. Photosynthetic nitrogen assimilation and associated carbon and respiratory metabolism. Springer Science & Business Media; 2006.
Yamasaki H. Nitrite–dependent nitric oxide production pathway: implications for involvement of active nitrogen species in photoinhibition in vivo. Philosophical Trans Royal Soc Lond Ser B: Biol Sci. 2000;355(1402):1477–88.
Willows RD, Li Y, Scheer H, Chen M. Structure of chlorophyll f. Org Lett. 2013;15(7):1588–90.
Coronel G, Chang M, Rodríguez-Delfín A, editors., editors. Nitrate reductase activity and chlorophyll content in lettuce plants grown hydroponically and organically. International Symposium on Soilless Culture and Hydroponics 843; 2008.
Reddy P, Ninganur BT, Chetti MB, Hiremath SM. Effect of growth retardants and nipping on chlorophyll content, nitrate reductase activity, seed protein content and yield in cowpea (Vigna unguiculata L). Karnataka J Agricultural Sci. 2009;22(2):289–92.
Gheibi MN, Malakouti MJ, Kholdebarin B, Ghanati F, Teimouri S, Sayadi R. Significance of nickel supply for growth and chlorophyll content of wheat supplied with urea or ammonium nitrate. J Plant Nutr. 2009;32(9):1440–50.
Wen B, Li C, Fu X, Li D, Li L, Chen X, et al. Effects of nitrate deficiency on nitrate assimilation and chlorophyll synthesis of detached apple leaves. Plant Physiol Biochem. 2019;142:363–71.
Aragao RM, Silva EN, Vieira CF, Silveira JAG. High supply of NO 3 – mitigates salinity effects through an enhancement in the efficiency of photosystem II and CO 2 assimilation in Jatropha curcas plants. Acta Physiol Plant. 2012;34:2135–43.
Nishimura K, Kato Y, Sakamoto W. Essentials of proteolytic machineries in chloroplasts. Mol Plant. 2017;10(1):4–19.
Murakami S, Kondo Y, Nakano T, Sato F. Protease activity of CND41, a chloroplast nucleoid DNA-binding protein, isolated from cultured tobacco cells. FEBS Lett. 2000;468(1):15–8.
Kato Y, Murakami S, Yamamoto Y, Chatani H, Kondo Y, Nakano T, et al. The DNA-binding protease, CND41, and the degradation of ribulose-1, 5-bisphosphate carboxylase/oxygenase in senescent leaves of tobacco. Planta. 2004;220:97–104.
Parsons DS, Volman-Mitchell H. The transamination of glutamate and aspartate during absorption in vitro by small intestine of chicken, guinea‐pig and rat. J Physiol. 1974;239(3):677–94.
Shim JS, Park S-H, Lee D-K, Kim YS, Park S-C, Redillas MCFR, et al. The rice GLYCINE-RICH PROTEIN 3 confers drought tolerance by regulating mRNA stability of ROS scavenging-related genes. Rice. 2021;14(1):1–19.
Kim JS, Jung HJ, Lee HJ, Kim KA, Goh CH, Woo Y, et al. Glycine-rich RNA‐binding protein7 affects abiotic stress responses by regulating stomata opening and closing in Arabidopsis thaliana. Plant J. 2008;55(3):455–66.
Gomez-Silva B, Timko MP, Schiff JA. Chlorophyll biosynthesis from glutamate or 5-aminolevulinate in intact Euglena chloroplasts. Planta. 1985;165:12–22.
Hendry GAF, Stobart AK. Glycine metabolism and chlorophyll synthesis in barley leaves. Phytochemistry. 1977;16(10):1567–70.
Wellburn FAM, Wellburn AR. Chlorophyll synthesis by isolated intact etioplasts. Biochem Biophys Res Commun. 1971;45(3):747–50.
Johnson AW, editor TRANSITION METAL COMPLEXES OF PORPHINS. Organometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic Chemistry; 2013: Elsevier.
Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303(5665):1831–8.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–D92.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15.
Zhu T, Wang L, Rimbert H, Rodriguez JC, Deal KR, De Oliveira R, et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese spring genome assembly. Plant J. 2021;107(1):303–14.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Li L, Liang X-F, He S, Sun J, Wen Z-Y, He Y-H, et al. Transcriptome analysis of grass carp (Ctenopharyngodon idella) fed with animal and plant diets. Gene. 2015;574(2):371–9.
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. The Innovation. 2021;2(3):100141.
Bathke J, Lühken G. OVarFlow: a resource optimized GATK 4 based Open source variant calling workFlow. BMC Bioinformatics. 2021;22:1–18.
Cingolani P. Variant annotation and functional prediction: SnpEff. Variant calling: methods and protocols. Springer; 2012. pp. 289–314.
Mia M, Liu H, Wang X, Zhang C, Yan G. Root transcriptome profiling of contrasting wheat genotypes provides an insight to their adaptive strategies to water deficit. Sci Rep. 2020;10(1):1–11.
Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M. Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol. 2009;10(1):1–27.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 – ∆∆CT method. Methods. 2001;25(4):402–8.
Acknowledgements
We would like to thank the Bioinformatics Center, Institute for Chemical Research, Kyoto University and Human Genome Center, Institute of Medical Science, University of Tokyo for developing KEGG software, and Kanehisa laboratories for providing the permission to publish under the CC BY 4.0 open-access license of KEGG pathway map images in this manuscript. We also thank Mrs. Anita Severn-Ellis for her support in molecular lab availability and induction and Mr. Greg Cawthray for technical support in operations of the Geno-grinder. We thank Novogene Singapore for next-generation sequencing services. The whole manuscript has been revised and edited by a professional English language editor, Dr Chris Davies of Tweak Editing.
Funding
This research was funded by the Global Innovation Linkages Project (GIL53853) from the Australian Department of Industry, Science and Resources, and The University of Western Australia.
Author information
Authors and Affiliations
Contributions
RB: conceptualisation, methodology, data curation, formal analysis, and writing – original draft. HL and GY: conceptualisation, funding acquisition, methodology, resources, supervision, and writing – review and editing. KHMS: supervision, and writing – review and editing. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The plant materials (the NILs) used in this study were developed at The University of Western Australia, with the parental cultivars Chuan Mai 25 and Ritchie obtained from Australian Grains Genebank under the Standard Material Transfer Agreement (AGG135560). The experimental research and plant material collection in this study comply with the institutional, national and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests for this research.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bhattarai, R., Liu, H., Siddique, K.H. et al. Transcriptomic profiling of near-isogenic lines reveals candidate genes for a significant locus conferring metribuzin resistance in wheat. BMC Plant Biol 23, 237 (2023). https://doi.org/10.1186/s12870-023-04166-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-023-04166-2