
Abstract
Background
Myrtales is a species rich branch of Rosidae, with many species having important economic, medicinal, and ornamental value. At present, although there are reports on the chloroplast structure of Myrtales, a comprehensive analysis of the chloroplast structure of Myrtales is lacking. Phylogenetic and divergence time estimates of Myrtales are mostly constructed by using chloroplast gene fragments, and the support for relationships is low. A more reliable method to reconstruct the species divergence time and phylogenetic relationships is by using whole chloroplast genomes. In this study, we comprehensively analyzed the structural characteristics of Myrtales chloroplasts, compared variation hotspots, and reconstructed the species differentiation time of Myrtales with four fossils and one secondary calibration point.
Results
A total of 92 chloroplast sequences of Myrtales, representing six families, 16 subfamilies and 78 genera, were obtained including nine newly sequenced chloroplasts by whole genome sequencing. Structural analyses showed that the chloroplasts range in size between 152,214–171,315 bp and exhibit a typical four part structure. The IR region is between 23,901–36,747 bp, with the large single copy region spanning 83,691–91,249 bp and the small single copy region spanning 11,150–19,703 bp. In total, 123–133 genes are present in the chloroplasts including 77–81 protein coding genes, four rRNA genes and 30–31 tRNA genes.
The GC content was 36.9–38.9%, with the average GC content being 37%. The GC content in the LSC, SSC and IR regions was 34.7–37.3%, 30.6–36.8% and 39.7–43.5%, respectively. By analyzing nucleotide polymorphism of the chloroplast, we propose 21 hypervariable regions as potential DNA barcode regions for Myrtales. Phylogenetic analyses showed that Myrtales and its corresponding families are monophyletic, with Combretaceae and the clade of Onagraceae + Lythraceae (BS = 100%, PP = 1) being sister groups. The results of molecular dating showed that the crown of Myrtales was most likely to be 104.90 Ma (95% HPD = 87.88–114.18 Ma), and differentiated from the Geraniales around 111.59 Ma (95% HPD = 95.50–118.62 Ma).
Conclusions
The chloroplast genome structure of Myrtales is similar to other angiosperms and has a typical four part structure. Due to the expansion and contraction of the IR region, the chloroplast genome sizes in this group are slightly different. The variation of noncoding regions of the chloroplast genome is larger than those of coding regions. Phylogenetic analysis showed that Combretaceae and Onagraceae + Lythraceae were well supported as sister groups. Molecular dating indicates that the Myrtales crown most likely originated during the Albian age of the Lower Cretaceous. These chloroplast genomes contribute to the study of genetic diversity and species evolution of Myrtales, while providing useful information for taxonomic and phylogenetic studies of Myrtales.
Similar content being viewed by others


Background
The Myrtales belong to the Rosidae, which is one of the most speciose groups in the Rosanae clade of angiosperms [1, 2]. According to APG IV [3], Myrtales consists of nine families, 380 genera, and approximately 13,000 species. The nine families in the order are Alzateaceae, Combretaceae, Crypteroniaceae, Lythraceae, Melastomataceae, Myrtaceae, Onagraceae, Penaeaceae and Vochysiaceae. The species richness of families is unbalanced with relatively few species found in Alzateaceae, Crypteroniaceae and Penaeaceae. Species are widely distributed in the tropics, with Vochysiaceae showing an amphi-Atlantic disjunct distribution [2]. Species in Combretaceae are mainly distributed in tropical and subtropical regions, especially in African savannahs [4]. The order is morphologically diverse with herbaceous herbs, lianas, trees, and mangroves, as well as a wide variety of fruit types (berry, capsule, samara and drupe) [1] (Fig. 1). There are two main wood anatomical characteristics of Myrtales: bilateral vascular bundles in the primary stem and vascular bundles in the marginal depressions of secondary xylem, which are not common in other flowering plants. The combination of these two anatomical characteristics is exceedingly rare [5,6,7]. Many of the species of Myrtales have important economic [8], ornamental [9] and medicinal value [10, 11].

Flowers of typical plants in six families of Myrtales
With the rapid development of second-generation sequencing technology, the cost of sequencing has made phylogenomic approaches feasible on large scales, ushering in a new exploration of plant identification and classification. Complete plastome sequences have become powerful tools to answer questions about plant evolution from inferred phylogenies [12,13,14,15,16,17,18]. The plastome is an essential organelle in photosynthetic cells, playing an important role in maintaining life [19] and is mainly maternally inherited in angiosperms. Most plastome DNA consist of double chains with a length of 120–220 kb [20] and a highly conserved typical four part genome structure. In recent years, researchers have been devoted to structural and phylogenetic analyses of chloroplasts in many groups, including Myrtales [21,22,23]. Structural characteristics of the chloroplasts have been useful for examining the genetic diversity and species evolution, and vital in developing policies for the protection of germplasm resources [24,25,26].
Reginato et al. [21] reported comparisons of chloroplast genomes in Melastomataceae for the first time. The structure, gene content and general characteristics of 16 chloroplast genomes of Melastomataceae and eight published chloroplast genomes of Myrtales were compared and analyzed. They found that the chloroplast genomes of Melastomataceae, like most angiosperms, have a typical tetrad structure with a large single copy region containing 84 protein coding genes (CDS), 37 tRNA and eight rRNA, for a total of 129 genes [21]. Gu et al. [22] reported the plastome of Heimia myrtifolia, an important medicinal plant with a variety of pharmacological alkaloids in the Lythraceae. Later, combined with 22 samples of other species in the Lythraceae, the chloroplast genome structure was comprehensively analyzed and compared with that of other species in Myrtales. The chloroplast genomes of 22 species of Lythraceae ranged from 152,049 bp to 160,769 bp, and included 10 variation hot spots that were selected as potential molecular markers [23]. In addition, other chloroplast genomes of Myrtales have been reported recently. Rodrigues et al. [27] compared the structure, gene number and genome size of six chloroplast genomes of Myrtales finding them to be similar to those of other Myrtales species. However, previous studies on chloroplast genomes of Myrtales have not been consistent, with some based on families, genera or species. Up to now, the comprehensive analysis of chloroplast genome structure of Myrtales is lacking.
In addition to studying the chloroplast genomes structure of Myrtales, researchers also explored the divergence time and phylogeny of Myrtales, but most studies were based on gene fragments. A strong phylogenetic framework is necessary to provide a basis for studying speciation. In previous molecular phylogenetic studies, a handful of chloroplast loci along with the internal transcribed spacer (ITS) and other ribosomal regions of nuclear DNA have been used for phylogenetic analysis of Myrtales [2, 7, 28]. Conti et al. [7] used 50 taxa (including 39 species and 11 outgroups) and the chloroplast gene rbcL to reconstruct the phylogeny of Myrtales. The results showed that Onagraceae and Lythraceae were closely related to Combretaceae [7]. Sytsma et al. [28] constructed the phylogenetic divergence time of Myrtales based on the chloroplast gene fragments rbcL and ndhF from 79 species of Myrtales and five fossil calibration points, indicating that Myrtales differentiated in the early Albian (111 Ma) with Combretaceae being the earliest branch of Myrtales with low support. Berger et al. [2] amplified and sequenced 6 gene fragments (rbcL, ndhF, matK, matR, 18S and 26S) from 102 taxa of Myrtales, and estimated the divergence time of Myrtales using 10 fossil calibration points. The results showed that the crown of Myrtales was most likely dated to 116 Ma (95% HPD = 113.7–118.8 Ma), while the phylogeny also showed that the Combretaceae is a sister group of all other families of Myrtales [2]. More recently, Li et al. [18] used 80 genes from 2881 plastomes and 62 fossil calibrations to reconstruct an angiosperm wide phylogeny showing that Myrtales and all of its families were monophyletic. The resulting phylogeny showed that the clade of Myrtales and Geraniales had a crown age of 112.26 Ma, as well as Combretaceae and Onagraceae + Lythraceae being sister groups with strong support. Most of the studies based on chloroplast gene fragments inferred relationships with low support, so using chloroplast genomes to explore the time of species differentiation and reconstruct phylogenetic relationship has credibility.
Currently there are few previous studies on the chloroplast genome structure of Myrtales. Although the phylogenetic position and relationships of Myrtales has been studied using molecular methods, the support for the placement of Myrtales is generally weak due to the lack of phylogenetic signal and sparse taxonomic sampling. Therefore, we set out to expand the sampling, reconstruct the phylogenetic relationship of Myrtales by using whole chloroplast genomes and comparatively analyze the plastome structure of Myrtales to provide the foundation for future research. In this study, we sequenced the chloroplast genomes of nine new species (including species of Myrtaceae, Melastomataceae and Combretaceae) and combined them with existing plastome data for Myrtales from NCBI to obtain a total of 95 chloroplast genomes, representing six families, 78 genera, and three outgroups. The main objectives of this study were to 1) analyze the chloroplast genome structure and elucidate the genetic diversity of Myrtales, 2) reconstruct the phylogenetic relationship of Myrtales to specifically determine the phylogenetic position of Combretaceae, and 3) infer the divergence time of Myrtales.
Results
Characteristics of chloroplast genomes
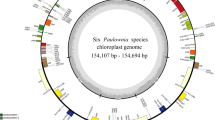
Six families were represented with the 92 Myrtales chloroplast genomes used in this study: Melastomataceae (42 species in five subfamilies), Myrtaceae (including 19 species in five subfamilies), Vochysiaceae (seven species), Lythraceae (13 species in three subfamilies), Onagraceae (three species in two subfamilies), and Combretaceae (eight species in one subfamily). All chloroplast genomes have a typical four part structure: large single copy region (LSC), small single copy region (SSC) and two inverted repeat regions (IRs) (Fig. 2). The length of the chloroplast genomes in the 42 samples of Melastomataceae ranged from 153,304 bp (Sarcopyramis napalensis, MK994868.1) to 157,991 bp (Astronia smilacifolia, MK994883.1), while the 19 samples of Myrtaceae ranged from 156,129 bp (Rhodomyrtus tomentosa, NC_043848.1) to 160,459 bp (Eucalyptus grandis). The chloroplast genomes of the seven Vochysiaceae samples ranged in length from 160,687 bp (Erisma bracteosum, NC_043794.1) to 171,315 bp (Vochysia acuminata, NC_043811.1), the 13 Lythraceae samples ranged from 152,214 bp (Lagerstroemia excelsa, NC_042896.1) to 160,054 bp (Pemphis acidula, NC_041439.1), and the three Onagraceae samples ranged from 159,396 bp (Ludwigia octovalvis, NC_031385.1) to 165,779 bp (Oenothera villaricae, NC_030532.1). Finally, the length of the chloroplast genomes in the eight samples of Combretaceae ranged from 159,750 bp (Terminalia guyanensis, NC_043807.1) to 161,773 bp (Combretum littoreum). Across all chloroplast genomes of Myrtales, the difference in plastome size between families was 19,101 bp, the difference of the IR region was 12,846 bp, the difference of the SSC region was 8553 bp, and the difference of the LSC region was 7558 bp. All 92 chloroplast genomes showed a typical quadripartite structure, comprising two IR regions (26,781–36,747 bp) separated by the LSC (83,691–91,249 bp) and the SSC (11,150–19,703 bp) regions (Table 1). In addition, a total of 123–133 genes are encoded, of which 106–116 are single copy with 17 genes duplicated in the IR regions. Of the unique genes 77–81 are protein coding genes, 29–31 are tRNA genes, and four are rRNA genes. The total GC content of the chloroplast genomes are highly similar (36.9–38.9%), with the average GC content across the entire chloroplast genomes being 37%, while the different regions had slightly variable GC content with the LSC, SSC and IR ranging from 34.7–37.3%, 30.6–36.8%, and 39.7–43.5%, respectively (Tables 1 and 2).

Chloroplast genome gene map of Myrtales. Genes on the inside of the outer circle are transcribed clockwise and those outsides are transcribed counterclockwise
Boundaries between IR and SC regions
In total, we analyzed and compared the differences between boundary regions of the SC and IR in 24 chloroplast genomes (15 samples from NCBI and the nine newly sequenced chloroplast genomes covering 16 subfamilies/families within Myrtales). We found that most chloroplast genomes have similar characteristics. The junction of the LSC/IRb region of 23 chloroplast genomes was located at the rps19 and rpl2 genes, while the junction of LSC/IRb region of Salpinga maranonensis (NC_031888.1) was unique with the boundary at the rpl2 gene. Except for Oenothera villaricae (NC_ 030532.1) the boundary of IRb/SSC was ccsA - ndhD. The ndhF gene was detected at the boundary of IRb/SSC in all other species. The ndhF gene of 11 species crossed the boundary of IRb/SSC, while ndhF of 12 species was completely found in the SSC region, ranging between 3 and 235 bp from the boundary. The gene ycf1 is at the SSC/IRa boundary except in Vochysia acuminata (NC_043811.1) and Oenothera villaricae (NC_030532.1). In total there are 20 species for which ycf1 crosses the boundary between SSC/IRa, two species in which ycf1 is completely in the SSC ranging from 63 to 381 bp away from the boundary, and one species in which ycf1 is completely in the IRa 1063 bp away from the boundary. The genes rpl2 and trnH (rpl2 is located in IRa, 53–139 bp away from the boundary, trnH is located in LSC, 0–216 bp away from the boundary) were detected in the IRa/LSC boundary for 20 species. The genes rps19 and trnH (rps19 is located in IRa, 0–3 bp away from the boundary, trnH is located in LSC, 1–41 bp away from the boundary) were detected in the IRa/LSC boundary for three species, and rpl23 and trnH were detected in the IRa/LSC boundary for Salpinga maranensis (NC_031888.1) (Fig. 3).

Comparison of the IR/SC junctions among 24 chloroplast genomes of Myrtales (15 samples from NCBI and the nine newly sequenced chloroplast genomes covering 16 subfamilies/families within Myrtales)
Comparative genomic analysis and divergence hotspot regions
We analyzed the comprehensive sequence divergence of the 24 Myrtales chloroplast genomes (15 samples from NCBI and the nine newly sequenced chloroplast genomes covering 16 subfamilies/families within Myrtales) using the mVISTA software with the annotation of V. acuminate used as a reference. A genome wide alignment revealed globally high sequence similarity (> 90% identity) (Fig. 4). The LSC and SSC regions show a higher level of sequence divergence than the inverted repeat regions. In addition, 188 regions were extracted to calculate nucleotide variability (Table S1). In coding regions, the loci with the largest variation are matK, rpoC2, accD, rpl20, ndhF, rpl32, ccsA, ndhD, and rps15; in non-coding regions, the loci with the largest variation are psbK-psbI, psbI-trnS (GCU), trnS (GCU)-trnG (GCC), trnR (UCU)-atpA, psbC-trnS (GCU), trnG-trnfM, trnF-ndhJ, ndhJ-ndhK, accD-psaI, rpl33-rps18, rps18-rpl20 and rps15-ycf1. DNA barcodes with the largest nucleotide diversity are considered to be the focus of phylogenetic analysis and plant identification (Fig. 5).

Visualization of the alignment of 24 chloroplast genome sequences of Myrtales. The plastome of Vochysia acuminata was used as the reference. The Y-axis depicts percent identity to the reference genome (50–100%) and the X-axis depicts sequence coordinates within the plastome. Genome regions were color-coded according to coding and non-coding regions

Comparison of the nucleotide diversity values across 92 chloroplast genomes of Myrtales. a Protein-coding regions. b Noncoding regions. The vertical dotted lines divides the approximate boundary of LSC, IRb and SSC
Phylogenetic results
Both ML and BI analyses of the complete chloroplast generated almost identical topologies with strong support at every node [ML bootstrap (BS) = 100%, Bayesian posterior probabilities (PP) = 1] (Fig. 6). Melastomataceae, Myrtaceae, Vochysiaceae, Onagraceae, Lythraceae, and Combretaceae were fully supported as monophyletic, with Combretaceae resolved as sister to Onagraceae + Lythraceae clade (BS/PP = 100/1; (Fig. 6). Melastomataceae was recovered as sister to Myrtaceae + Vochysiaceae (BS/PP = 100/1). A clade of Melastomataceae + Myrtaceae + Vochysiaceae was recovered as sister to the clade of Combretaceae + Onagraceae + Lythraceae with strong support (BS/PP = 100/1). In addition, the phylogenetic trees constructed using the coding regions (CR), noncoding regions (NCR), LSC, SSC and NO-IRa phylogenetic trees (ML / BI) have the same topological structure at the family level as the phylogeny inferred from the full chloroplast with strong support (Figure S1, S2, S3. S4 and S5). Observed differences were found in the phylogenetic relationships constructed by the IRb region, in which Melastomataceae was resolved as sister to Myrtaceae + Vochysiaceae + Lythraceae + Combretaceae, and Lythraceae was resolved as a sister to Combretaceae albeit with low support (Figure S6). Additionally, we expanded the outgroups to construct the phylogenetic relationship of Malvids, and the phylogenetic relationship of Myrtales was also strongly supported (Figure S7).

Optimal phylogenetic tree resulting from analyses of 92 complete chloroplast genomes of Myrtales and 3 outgroups using Maximum Likelihood (ML) and Bayesian inference (BI). Support values are maximum likelihood bootstrap support/Bayesian posterior probability; asterisks indicate 100%/1.0 support values. The families of Myrtales are indicated by different colors. The inset shows the same tree as a phylogram
Divergence time estimation of Myrtales
The results of the BEAST analysis of species divergence time in Myrtales are shown in Fig. 7. The crown age of Myrtales is 104.90 Ma (95% HPD = 87.88–114.18 Ma) with the recent common ancestor with Geraniales dated to 111.59 Ma (95% HPD = 95.50–118.62 Ma) during the Albian age of the Lower Cretaceous. Based on the BEAST chronogram, the Combretaceae with Onagraceae + Lythraceae (crown group age: 89.59 Ma, HPD = 81.02-108.93 Ma) diverged 96.22 Ma (95% HPD = 81.03–109.26 Ma) in the Cenomanian age of the Upper Cretaceous. The crown group of Melastomataceae (crown group age: 45.82 Ma, 95% HPD = 13.72–71.50 Ma) with Myrtaceae + Vochysiaceae (crown group age: 86.43 Ma, 95% HPD = 83.52–106.94 Ma) diverged at 94.21 Ma (95% HPD = 83.54–106.94 Ma) in the Cenomanian age of the Upper Cretaceous.

Chronogram of Myrtales based on complete chloroplast genomes sequences estimated from BEAST. The blue circle represents four fossil constraints and one grey circle represents secondary constraint, and the yellow boxes represent our estimated divergence times of major lineages
Discussion
Plastome structure comparisons and sequence divergence hotspots
Previous studies have shown that the size of chloroplast genomes in angiosperms are between 120 and 180 kb, and the size of IR region is 20–30 kb [29]. The size range of the 92 chloroplast genomes in Myrtales is 152,214–171,315 bp, of which the IR is 26,781–36,747 bp. Our results show that the chloroplast genomes of Myrtales are on the larger end of organellar genomes in angiosperms. The largest plastome is in the Vochysiaceae, and the smallest plastome is in the Lythraceae. The difference of plastome length between different families mainly lies in the difference of IR region length. The change in the overall length of chloroplast genomes is generally related to the expansion and contraction of IR regions [30]. The presented results are similar to those found in Pelargonium hortorum, Cryptomeria fortunei, Geranium, Pisum sativum, Vicia faba, and Erodium in which the size of the IR is increased, decreased or even completely lost [31,32,33,34]. In angiosperms, high conservation of the IR region is common, and is important for stabilizing plastome gene structure [35] though changes have been reported including in some early diverging eudicots [36, 37].
The nucleotide content of chloroplasts is relatively stable and the gene structure is highly conserved, though mutation hotspots do exist. Genes with a relatively high mutation rate can be used as DNA barcodes to help distinguish between accessions within a given taxon [38, 39] and varieties in germplasm resources [40, 41]. In this study, we used mVISTA to compare the whole chloroplast of 24 species of Myrtales and used DnaSP to analyze the percentage of variable loci in 74 coding genes and 114 non-coding regions. Similar to previous results, the variation of noncoding regions is greater than that of coding regions [42, 43]. As observed in members of Adoxaceae and Panax notoginseng, the variation of the IR region of Myrtales is smaller than that of the SC region [44, 45]. Previous studies investigating the phylogeny of Myrtales using only rbcL failed to resolve the phylogenetic position of the order. Our analyses showed that the nucleotide diversity of rbcL is relatively low compared to other loci (PI < 0.05) (Fig. 5, Table S1), which helps explain the low support found in phylogenies inferred with this gene [7]. We detected nine hot spots in coding regions and 12 hot spots in noncoding regions, which can be used as candidate DNA barcodes for future studies. These variable regions may also be useful for assessing phylogenetic relationships and interspecific differences of Myrtales species.
Phylogenetic relationships of Myrtales
Compared with previous studies based on a few chloroplast genome fragments, our results based on the major lineages of Myrtales (six families with more species within Myrtales) showed a highly resolved phylogenetic relationship of Myrtales by using whole chloroplast genomes [2, 6, 28]. Six major clades representing the major families are fully resolved with strong support (Fig. 7). Previous studies of Myrtales have provided an improved understanding of phylogenetic relationships among families based on both morphological and molecular analyses, however, the placement of Combretaceae has not been fully established with high confidence [2, 6, 28]. The phylogenetic location of Combretaceae is critical since its placement directly affects the age of Myrtales, hypotheses of diffusion and variation scenarios, species diversification rates, and features of trait reconstructions [2]. Most recent phylogenetic studies use a limited number of taxa and gene regions as placeholders for Combretaceae [7, 28, 46, 47]. Our plastome phylogenomic analysis of Myrtales provides strong support for the sister relationship between Combretaceae and a clade of Onagraceae + Lythraceae (BS = 100%, PP = 1; Fig. 7), which is in agreement with some previous molecular studies, and a clade of Combretaceae + Onagraceae + Lythraceae is sister to a clade of Melastomataceae + Myrtaceae + Vochysiaceae [18, 48]. The sampling of our study is not comprehensive at the family level with the phylogenetic relationship reconstructed including six of the nine families (lack samples from Crypteroniaceae, Penaeaceae and Alzateaceae). However, according to previous studies, this does not affect our determination of the phylogenetic position of the Combretaceae. We used the whole chloroplast genome to construct the phylogenetic relationships, as well as using multiple chloroplast gene data sets (excluding the chloroplast genome of IRa region, coding genes, noncoding genes, LSC, SSC, IRb) to compare the phylogenetic relationship comprehensively. We also reconstructed the phylogenetic relationship by adding extra taxa (within the branch of Malvids), providing an additional degree of credibility for the obtained phylogenetic trees [49, 50] and determining the phylogenetic position of the Combretaceae. Further research should include sampling more individuals from wild populations and obtaining more extensive nuclear data to determine whether our results are consistent with those from nuclear genes.
Molecular dating
Biogeography estimates generally suggested that the Myrtales originated in Gondwana [7, 28, 46, 51, 52] with the diversity of all major stem lineages being traced to 85–90 Ma in the western portion of Gondwana. The results of the molecular dating showed that the crown group of Myrtales most likely originated in the Albian age of the Lower Cretaceous [104.90 Ma (95% HPD = 87.88–114.18 Ma)]. The estimated divergence time of Myrtales (Fig. 6) presented here is in close proximity to previously reported dates (104.90 Ma compared to 111 Ma, Sytsma et al. [28]; 116.4 Ma, Berger et al. [2]; 90.7 Ma, Thornhill et al. [53]). However, Gonçalves et al. [54] using 78 protein coding genes from 122 chloroplast genomes of Myrtales, combined with four Myrtales fossil sites and a secondary calibration point, estimated the divergence time of Myrtales to be 125.5 Ma (95% HPD = 130.9–120.3 Ma) during the upper Cretaceous. Fossil limitations, different methods, size of molecular data and taxonomic sampling cannot be perfectly compared across all studies, with changes leading to differences in age estimates. Our analysis estimated that the diversity of major lineages of Myrtales occurred about 60–90 Ma [2, 18]. In this period the species within Myrtales may have begun to differentiate rapidly, which is consistent with the common hypothesis that many species experienced rapid diversification events after the Cretaceous-Paleogene (K-Pg) boundary due to mass extinction and opening of new habitats [55,56,57]. Our results show that the species diversity of the main stem lineages of Myrtales increased at the end of the Campanian and may have been affected by the continental breakup of Gondwana in the Cretaceous [2].
Conclusions
In this study, we analyzed and compared the structural characteristics of chloroplast genomes of Myrtales, and inferred the phylogenetic divergence time of Myrtales. The chloroplast genomes of Myrtales has a typical four part structure, including 77–81 protein coding genes, 29–31 tRNA genes and four rRNA genes, with a total length of 152,214–171,315 bp. We found 21 mutation hotspots, which can be used as potential DNA barcodes in the future phylogenetic study of Myrtales. Phylogenetic relationships (Ml / BI) based on whole chloroplast genome and multiple datasets showed that Myrtales and its families were monophyletic, as well as Combretaceae and Onagraceae + Lythraceae strongly supported as a clade, (BS = 100%, PP = 1). Reconstructing the divergence time of Myrtales shows that the crown of Myrtales is 104.90 Ma (95% HPD = 87.88–114.18 Ma), and it differentiated from Geraniales around 111.59 MA (95% HPD = 95.50–118.62 MA) in the Albian of the early Cretaceous. The species divergence of Myrtales ranged from 60 to 90 Ma. These chloroplast genomes contribute to the study of genetic diversity and species evolution of Myrtales, while providing useful information for taxonomic and phylogenetic studies of Myrtales. In the future, we will expand genomic sampling, including nuclear genomes, to comprehensively compare and discuss the phylogeny and evolution of Myrtales species.
Methods
Taxon sampling
Leaf material from nine species, representing seven genera and three families in Myrtales, was collected and stored in silica gel. Combretum kraussii Hochst., Eucalyptus grandis W. Mill ex Maiden, Melaleuca leucadendra Linn., Combretum littoreum (Engl.) Exell, Syzygium forrestii Merr. et Perry, S. cumini (Linn.) Skeels and Tibouchina semidecandra Cogn. were collected from the Ruili Botanical Garden (Yunnan Province, China; 23°52′ to 24°09′ E, 97°38′ to 98°05′ N). Combretum malabaricum Linn. and Terminalia catappa Linn. were collected from Hainan University (Hainan province of China; 20°05′ to 20°06′ E, 110°33′ to 110°34′ N). The sampling of nine newly sequenced species was approved by Ruili Botanical Garden (Yunnan Province, China) and Hainan University (Hainan province of China) and met local policy requirements. Table 3 indicates the detailed voucher and locality information for the newly sequenced species. In addition, 83 species representing six families of Myrtales and three outgroups (Viviania marifolia, NC_023259.1; Pelargonium tetragonum, NC_031205.1; Pelargonium quercifolium, NC_031203.1) were downloaded from NCBI with detailed information presented in Table 1. We also downloaded 17 chloroplast genomes from NCBI, including six different orders to serve as outgroups to construct a branch of Malvids to explore the topological changes of Myrtales (Table S2).
DNA extraction, sequencing and assembly
We used a modified cetyltrimethyl ammonium bromide (CTAB) method to extract high quality DNA from dried leaves [58]. Quality of DNA was determined on an Agilent 2100 BioAnalyzer by using ≥0.8 μg at the University of California Davis Genome Center (Davis, California, USA). We constructed paired-end sequencing libraries with insert sizes of 200–400 bp with Illumina TruSeq™ Nano DNA Sample Prep Kit and sequenced using the BGISEQ-500 at the Beijing Genomics Institution (BGI; Shenzhen, China). Raw reads were filtered with SOAPfilter_v2.2 for quality control with the following parameters: 1) remove low quality reads (> 10% Ns and/or > 40% low quality bases), 2) remove PCR duplicates, and 3) trim adaptor sequences. We selected the rbcL gene of Arabidopsis thaliana from NCBI (accession number: U91966) as a seed and assembled chloroplast genomes for each species using the clean reads with NOVOPlasty [59]. The longest contig assembled by NOVOPlasty was compared with chloroplasts deposited in the NCBI database, and obtained the chloroplast genome sequence with the highest homology (minimum requirement: e-value < 10–7, identity > 95%) to us as the reference (Table 3) for subsequent assembly using MITObim v1.8 [60]. Quality of the assemblies were assessed by mapping clean reads using BWA MEM (Burrows-Wheeler Aligner) v0.7.17 [61] to verify the integrity of newly assembled plastome [62].
Plastome annotation
Plastome sequences were initially annotated using Geneious R11.0.4 (Biomatters Ltd., Auckland, New Zealand), then further annotated with Dual Organellar GenoMe Annotator (DOGMA) [63] to modify gene boundaries. The tRNA genes were verified with tRNAscan-SE1.21 [64]. Maps were drawn using OrganellarGenomeDRAW v1.3.1 (available online: https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) [65] (Fig. 3). All plastome sequences have been uploaded to NCBI (Table 3).
Plastome comparative analysis and molecular marker identification
Plastome comparisons across 24 Myrtales species (15 samples from NCBI and the nine newly sequenced chloroplast genomes covering 16 subfamilies/families within Myrtales) were performed in Shuffle-LAGAN mode on the mVISTA program (genome.lbl.gov/vista/index.shtml [66];), using the annotation of Vochysia acuminate (NC_043811) as a reference. To reveal highly variable regions for future species identification studies and to evaluate different plastome regions that may show different evolutionary patterns, we sequentially extracted both coding regions and noncoding regions (including intergenic spacers and introns) after alignment with MAFFT v7 [67] using the criteria that the aligned length is > 200 bp and at least one mutation per site was present. The nucleotide variability of the selected regions was evaluated using DNASP v5.10 [68]. The IR / SC boundary map of these 24 Myrtales chloroplast was drawn with Photoshop. The IR area was confirmed using UNIPRO ugene v1.32 [69].
Phylogenetic analysis
Phylogenetic analyses were conducted on 95 species, using Viviania marifolia (NC_023259), Pelargonium tetragonum (NC_031205), and Pelargonium quercifolium (NC_031203) as outgroups based on a previous study [2]. Plastome sequences were aligned using MAFFT v7 [67] and manually checked when necessary. The complete chloroplast genome sequence and chloroplast genome minus one copy of the inverted repeat (No-IRa) were used to construct the phylogenetic topology using maximum likelihood (ML) and Bayesian inference (BI). To evaluate alternative hypotheses, phylogenetic topologies were inferred using both maximum likelihood (ML) and Bayesian inference (BI) methods using the complete plastome sequences and whole plastome minus one copy of the Inverted Repeat (No-IRa). We also included other data sets (i.e., coding area, noncoding area, LSC, SSC and IRb) for analyses. The best-fitting model of molecular evolution (GTR + GAMMA+I) (Table 4) was determined using Akaike Information Criterion (AIC) in JMODELTEST v2.1.7 [70]. Maximum likelihood analyses were conducted in RAxML-HPC v8.2.8 [71] with 1000 bootstrap replicates on the CIPRES Science Gateway portal [72]. Bayesian analyses were performed in MRBAYES v3.2 [73]. Two independent Markov Chain Monte Carlo chains were conducted simultaneously for 5 million generations with trees sampled every 1000 generations. The effective sample size (ESS > 200) was determined using Tracer v1.7 [74] and the first 25% of trees were discarded as burn-in, and a consensus tree was constructed from the remaining trees to estimate posterior probabilities (PPs). FigTree v1.4.4 [75] were used for visualizing the resulting phylogenetic trees.
Divergence time estimation
The complete 92 plastome dataset of Myrtales was analyzed using the GTR + GAMMA+I model selected by MrModelTest [76] in BEAST v.1.8.4 [75] to simultaneously search for the best tree topology and estimate node ages. The divergence time between lineages was estimated using a Yule speciation prior and an uncorrelated lognormal model of rate change with a relaxed clock. Four fossil-based calibration points and one secondary calibration point were used to constrain the crown node age of Myrtales. (1) The Myrtaceidites (=Syncolporites) pollen [28] placed a prior on the crown of Myrtaceae. The Myrtaceidites lisamae (83.5 Ma) fossil from Gabon, Africa during the Santonian [52, 77, 78] was considered the oldest fossil in Myrtaceae. Therefore, we set the stem of Myrtaceae with a lognormal mean = 0, a SD = 1.0 and an offset = 83.5 Ma. (2) In the Chamelaucioideae clade of Myrtaceae we placed the fossil of Eucalyptus frenguelliana (51.69 Ma) dated to the early Eocene from Laguna del Hunco in Chubut Province, Argentina [79, 80]. We set the stem of Chamelaucioideae with a lognormal mean = 0, a SD = 1.0 and an offset = 51.69 Ma. (3) The stem of Lythraceae was set to a lognormal mean = 0, a SD = 1.0 and an offset = 81.0 Ma based on the pollen fossil for Lythrum elkensis of Lythrum/Peplis from the Late Cretaceous (early Campanian, 82–81 Ma) in Wyoming, USA [80, 81]. (4) We used the earliest recorded wood fossil of Sonneratioxylon preapetalum Awasthi [82] from the early Paleocene in India (Danian, 67.3–63.8 Ma) [81] to constrain the node of Trapoideae. We set the stem to 63.8 Ma with a lognormal mean equal to 0 and a standard deviation of 1. (5) Based on the results of Li et al. [18], the clade of Myrtales and Cerambycidales had a crown age of 112.26 Ma, the crown node age of Myrtales+Geraniales was constrained to 112.26 Ma, with a normal prior and SD = 5. Nine runs each with 100 million generations were conducted totaling 900 million generations with parameters sampled every 1000 generations. The effective sample size (> 200) was determined using Tracer v1.6 [75] and the first 25% of the samples were discarded as burn-in. TreeAnnotator v1.8.0 [75] was used to produce a maximum clade credibility chronogram showing the mean divergence time estimates with 95% highest posterior density (HPD) intervals. FigTree v1.4.4 [75] was used to visualize the resulting divergence times.
Availability of data and materials
All sequences used in this study are available from the National Center for Biotechnology Information (NCBI) (accession numbers: MT700492- MT700490; see Additional Table 2).
Abbreviations
- BI:
-
Bayesian Inference
- CTAB:
-
Cetyltrimethylammonium bromide
- DnaSP:
-
DNA Sequences Polymorphism
- IR:
-
Inverted repeat
- LSC:
-
Large single copy
- GTR:
-
General time reversible
- ML:
-
Maximum Likelihood
- PI:
-
Phylogenetic informativeness
- rRNA:
-
Ribosomal RNA
- SSC:
-
Small single copy
- tRNA:
-
Transfer RNA
References
Dahlgren R, Thorne R. The order Myrtales: circumscription, variation, and relationships. Ann Mo Bot Gard. 1984;71(3):633–99. https://doi.org/10.2307/2399158.
Berger BA, Kriebel R, Spalink D, Sytsma KJ. Divergence times, historical biogeography, and shifts in speciation rates of Myrtales. Mol Phylogenet Evol. 2016;95:116–36. https://doi.org/10.1016/j.ympev.2015.10.001.
Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botan J Linnean Soc. 2016;181:1–20.
Tan F, Shi S, Yang Z, Xun G, Wang Y. Phylogenetic relationships of Combretoideae (Combretaceae) inferred from plastid, nuclear gene and spacer sequences. J Plant Res. 2002;115(6):475–81. https://doi.org/10.1007/s10265-002-0059-1.
Van Vliet GJ, Baas P. Wood anatomy and classification of the Myrtales. Ann Mo Bot Gard. 1984;71(3):783–800. https://doi.org/10.2307/2399162.
Conti E, Litt A, Sytsma KJ. Circumscription of Myrtales and their relationships to other rosids: evidence from rbcL sequence data. Am J Bot. 1996;83(2):221–33. https://doi.org/10.1002/j.1537-2197.1996.tb12700.x.
Conti E, Litt A, Wilson PG, Graham SA, Briggs BG, Johnson L, et al. Interfamilial relationships in Myrtales: molecular phylogeny and patterns of morphological evolution. Syst Bot. 1997;22(4):629–47. https://doi.org/10.2307/2419432.
Thornhill AH, Ho SY, Külheim C, Crisp MD. Interpreting the modern distribution of Myrtaceae using a dated molecular phylogeny. Mol Phylogenet Evol. 2015;93:29–43. https://doi.org/10.1016/j.ympev.2015.07.007.
Peng DH, Zhang QX, Huang JT. Melastomataceae ornamental plant Germplasm resources in China and the distribution investigation in Fujian Province. Chin Landscape Architect. 2007;11:92–7.
Granato D, Nunes DS, Barba FJ. An integrated strategy between food chemistry, biology, nutrition, pharmacology, and statistics in the development of functional foods: A proposal. Trends Food Ence Technol. 2017;62(Complete):13–22.
Yoshida T, Amakura Y, Yoshimura M. Structural features and biological properties of ellagitannins in some plant families of the order Myrtales. Int J Mol Sci. 2010;11(1):79–106. https://doi.org/10.3390/ijms11010079.
Jansen RK, Cai Z, Raubeson LA, Daniell H, de Pamphilis CW, Leebens-Mack J, et al. Analysis of 81 genes from 64 plastome genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci. 2007;104:19369–74.
Moore MJ, Dhingra A, Soltis PS, Shaw R, Farmerie WG, Folta KM, et al. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2010;6:1–13.
Yang Y, Zhou T, Duan D, Yang J, Feng L, Zhao G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front Plant Sci. 2016;7:573–5.
Lu R-S, Li P, Qiu Y-X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front Plant Sci. 2017;7:2054.
Niu YT, Florian J, Barrett RL, Ye JF, Zhang ZZ, Lu KQ, et al. Combining complete chloroplast genome sequences with target loci data and morphology to resolve species limits in Triplostegia (Caprifoliaceae). Mol Phylogenet Evol. 2018;129:15–26. https://doi.org/10.1016/j.ympev.2018.07.013.
Pinard D, Myburg AA, Mizrachi E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genomics. 2019;20:1471–2164.
Li HT, Yi TS, Gao LM, Ma PF, Zhang T, Yang JB, et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants. 2019;5(5):461–70. https://doi.org/10.1038/s41477-019-0421-0.
Xiong AS, Peng RH, Zhuang J, Gao F, Zhu B, Fu XY, et al. Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnol Adv. 2009;27(4):340–7. https://doi.org/10.1016/j.biotechadv.2009.01.012.
Rogalski M, do Nascimento Vieira L, Fraga HP, Guerra MP. Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front Plant Sci. 2015;6:586.
Reginato M, Neubig KM, Majure LC, Michelangeli FA. The first complete plastid genomes of Melastomataceae are highly structurally conserved. Peer J. 2016;4:e2715. https://doi.org/10.7717/peerj.2715.
Gu C, Dong B, Xu L, Tembrock L, Zheng S, Wu Z. The complete chloroplast genome of Heimia myrtifolia and comparative analysis within myrtales. Molecules. 2018;23(4):846. https://doi.org/10.3390/molecules23040846.
Gu C, Ma L, Wu Z, Chen K, Wang Y. Comparative analyses of chloroplast genomes from 22 Lythraceae species: inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biol. 2019;19(1):281. https://doi.org/10.1186/s12870-019-1870-3.
Lin W, Huang J, Xue M, et al. Characterization of the complete chloroplast genome of Chinese rose, Rosa chinensis (Rosaceae: Rosa). Mitochondrial DNA Part B Resour. 2019;4(2):2984–5.
Xue ZQ, Xue JH, Victorovna M, Ma KP. The complete chloroplast DNA sequence of Trapa maximowiczii Korsh (Trapaceae), and comparative analysis with other Myrtales species. Aquat Bot. 2017;143:54–62. https://doi.org/10.1016/j.aquabot.2017.09.003.
Yang JY, Pak JH, Kim SC. The complete plastome sequence of Rubus takesimensis endemic to Ulleung Island, Korea: insights into molecular evolution of anagenetically derived species in Rubus (Rosaceae). Gene. 2018;668:221–8. https://doi.org/10.1016/j.gene.2018.05.071.
Rodrigues NF, Balbinott N, Paim I, et al. Comparative analysis of the complete chloroplast genomes from six Neotropical species of Myrteae (Myrtaceae). Genet Mol Biol. 2020;43(2):e20190302.
Sytsma KJ, Litt A, Zjhra ML, Chris Pires J, Nepokroeff M, Conti E, et al. Clades, clocks, and continents: historical and biogeographical analysis of Myrtaceae, Vochysiaceae, and relatives in the southern hemisphere. Int J Plant Sci. 2004;165(S4):S85–S105. https://doi.org/10.1086/421066.
Zhang T, Fang Y, Wang X, Deng X, Zhang X, Hu S, et al. The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: insights into the evolution of plant organellar genomes. PLoS One. 2012;7(1):e30531. https://doi.org/10.1371/journal.pone.0030531.
Wang W, Messing J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS One. 2011;6(9):e24670. https://doi.org/10.1371/journal.pone.0024670.
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, et al. The complete chloroplast genome sequence of Pelargonium× hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 2006;23(11):2175–90. https://doi.org/10.1093/molbev/msl089.
Guisinger MM, Kuehl JV, Boore JL, Jansen RK. Extreme reconFigureuration of chloroplast in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol. 2011;28(1):583–600. https://doi.org/10.1093/molbev/msq229.
Hirao T, Watanabe A, Kurita M, Kondo T, Takata K. Complete nucleotide sequence of the Cryptoeria japonica D. Don. Chloroplast genome and comparative chloroplast genomics: diversified genomic structure of coniferous species. BMC Plant Biol. 2008;8(1):1–20.
Hu YJ. Plastome genome structure and plastome genes. Plant Physiol Commun. 1985;2:65–71.
Maréchal A, Brisson N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010;186(2):299–317. https://doi.org/10.1111/j.1469-8137.2010.03195.x.
Downie SR, Jansen RK. A comparative analysis of whole plastome from the Apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst Bot. 2015;40(1):336–51. https://doi.org/10.1600/036364415X686620.
Sun Y, Moore MJ, Zhang S, Soltis PS, Soltis DE, Zhao T, et al. Phylogenomic and structural analyses of 18 complete chloroplast across nearly all families of early-diverging eudicots, including an angiosperm-wide analysis of IR gene content evolution. Mol Phylogenet Evol. 2016;96:93–101. https://doi.org/10.1016/j.ympev.2015.12.006.
Kuang DY, Wu H, Wang YL, Gao LM, Zhang SZ, Lu L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 2011;54(8):663–73. https://doi.org/10.1139/g11-026.
Mehmood F, Shahzadi I, Waseem S, Mirza B, Ahmed I, Waheed MT. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics. 2020;112(1):581–91.
Ge Y, Dong X, Wu B, Wang N, Chen D, Chen H, et al. Evolutionary analysis of six chloroplast genomes from three Persea americana ecological races: insights into sequence divergences and phylogenetic relationships. PLoS One. 2019;14(9):e0221827. https://doi.org/10.1371/journal.pone.0221827.
Zhou T, Wang J, Jia Y, Li W, Xu F, Wang X. Comparative chloroplast genome analyses of species in Gentiana section Cruciata (Gentianaceae) and the development of authentication markers. Int J Mol Sci. 2018;19(7):1962. https://doi.org/10.3390/ijms19071962.
Perry AS, Wolfe KH. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J Mol Evol. 2002;55(5):501–8. https://doi.org/10.1007/s00239-002-2333-y.
Huang H, Shi C, Liu Y, Mao SY, Gao LZ. Thirteen Camelliachloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14(1):151.
Fan WB, Wu Y, Yang J, Shahzad K, Li ZH. Comparative chloroplast genomics of dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front Plant Sci. 2018;9:689. https://doi.org/10.3389/fpls.2018.00689.
Dong W, Liu H, Xu C, et al. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 2014;15(1):1–8.
Johnson LAS, Briggs BG. Myrtales and Myrtaceae-a phylogenetic analysis. Ann Mo Bot Gard. 1984;71(3):700–56. https://doi.org/10.2307/2399159.
Magallón S. Using fossils to break long branches in molecular dating: a comparison of relaxed clocks applied to the origin of angiosperms. Syst Biol. 2010;59(4):384–99. https://doi.org/10.1093/sysbio/syq027.
Wang XQ, Song WW, Xiao JJ. Phylogeny of Myrtales and related groups based on chloroplast genome. Guihaia Plants. 2021;41:68–80. https://doi.org/10.11931/guihaia.gxzw201906024.
Smith SA, Beaulieu JM, Donoghue MJ. Mega-phylogeny approach for comparative biology: an alternative to supertree and supermatrix approaches. BMC Evol Biol. 2009;9(1):1–12.
Sanderson MJ, McMahon MM, Steel M. Phylogenomics with incomplete taxon coverage: the limits to inference. BMC Evol Biol. 2010;10(1):1–13.
Rutschmann F, Eriksson T, Salim KA, Conti E. Assessing calibration uncertainty in molecular dating: the assignment of fossils to alternative calibration points. Syst Biol. 2007;56(4):591–608. https://doi.org/10.1080/10635150701491156.
Muller J. Fossil pollen records of extant angiosperms. Bot Rev. 1981;47(1):1–142. https://doi.org/10.1007/BF02860537.
Thornhill AH, Popple LW, Carter RJ, Ho SYW, Crisp MD. Are pollen fossils useful for calibrating relaxed molecular clock dating of phylogenies? A comparative study using Myrtaceae. Mol Phylogenet Evol. 2012;63(1):15–27. https://doi.org/10.1016/j.ympev.2011.12.003.
Gonçalves DJP, Shimizu GH, Ortiz EM, Jansen RK, Simpson BB. Historical biogeography of Vochysiaceae reveals an unexpected perspective of plant evolution in the Neotropics. Am J Bot. 2020;107(7):1004–20. https://doi.org/10.1002/ajb2.1502.
Jablonski D. Mass extinctions and macroevolution. Paleobiology. 2005;31(sp5):192–210. https://doi.org/10.1666/0094-8373(2005)031[0192:MEAM]2.0.CO;2.
Schulte P, Alegret L, Arenillas I, Arz JA, Barton PJ, Bown PR, et al. The Chicxulub asteroid impact and mass extinction at the cretaceous-Paleogene boundary. Science. 2010;327(5970):1214–8. https://doi.org/10.1126/science.1177265.
Zhai W, Duan X, Zhang R, Guo C, Li L, Xu G, et al. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol Phylogenet Evol. 2019;135:12–21. https://doi.org/10.1016/j.ympev.2019.02.024.
Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue [R], vol. 19; 1987. p. 11–5.
Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4):e18.
Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41(13):e129. https://doi.org/10.1093/nar/gkt371.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv. 2013;1303:3997.
Liu H, Wei J, Yang T, et al. Molecular digitization of a botanical garden: high-depth whole-genome sequencing of 689 vascular plant species from the Ruili Botanical Garden. GigaScience. 2019;8(4):giz007.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–5. https://doi.org/10.1093/bioinformatics/bth352.
Peter S, Angela NB, Todd ML. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:686–9.
Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41(W1):W575–81. https://doi.org/10.1093/nar/gkt289.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32(suppl_2):W273–9.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. https://doi.org/10.1093/molbev/mst010.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. https://doi.org/10.1093/bioinformatics/btp187.
Rose R, Golosova O, Sukhomlinov D, Tiunov A, Prosperi M. Flexible design of multiple metagenomics classification pipelines with UGENE. Bioinformatics. 2019;35(11):1963–5. https://doi.org/10.1093/bioinformatics/bty901.
Santorum JM, Darriba D, Taboada GL, Posada D. Jmodeltest. Org, selection of nucleotide substitution models on the cloud. Bioinformatics. 2014;30(9):1310–1. https://doi.org/10.1093/bioinformatics/btu032.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. https://doi.org/10.1093/bioinformatics/btu033.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway forinference of large phylogenetic trees. In: Gateway Computing Environments Workshop; 2010. p. 1–8.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–4. https://doi.org/10.1093/bioinformatics/btg180.
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarisation in Bayesian phylogenetics using tracer 1.7. Syst Biol. 2018;67(5):901–4. https://doi.org/10.1093/sysbio/syy032.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73. https://doi.org/10.1093/molbev/mss075.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25(7):1253–6. https://doi.org/10.1093/molbev/msn083.
Boltenhagen E. Pollens et Spores Senoniens du Gabon. Cahiers Micropaleontol. 1976;3:1–21.
Herngreen GFW. An upper Senonian pollen assemblage of borehole 3-PIA-10-AL state of Alagoas, Brazil. Pollen Spores. 1975;17:93–140.
Gandolfo MA, Hermsen EJ, Zamaloa MC, Nixon KC, González CC, Wilf P, et al. Oldest known Eucalyptus macrofossils are from South America. PLoS One. 2011;6(6):e21084. https://doi.org/10.1371/journal.pone.0021084.
Grímsson F, Zetter R, Hofmann CC. Lythrum and Peplis from the late cretaceous and Cenozoic of North America and Eurasia: new evidence suggesting early diversifification within the Lythraceae. Am J Bot. 2011;98(11):1801–15. https://doi.org/10.3732/ajb.1100204.
Graham SA. Fossil records in the Lythraceae. Bot Rev. 2013;28:410–20.
Awasthi N. A fossil wood of Sonneratia from the tertiary of South India. Palaeobotanist. 1968;17:254–7.
Acknowledgements
We would like to thank anonymous reviewers for their thoughtful comments and constructive suggestions towards improving our manuscript.
Funding
This research was funded by a start-up fund from Hainan University (kyqd1633). The cost of sample collection and sequencing analysis was funded by this funding source.
Author information
Authors and Affiliations
Contributions
XFZ performed all experiments, analyzed the data and wrote manuscript. HXW and ZXZ assisted with the experiments. JBL help to revise the manuscript. HFW planned and directed the study and revised the manuscript. It is to mention that all authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figures S1–S6.
are phylogenetic relationships inferred by Maximum Likelihood and Bayesian inference based on: coding genes; noncoding loci; the LSC (the Large Single-Copy); the SSC (the Small Single-Copy); NO-IRa data set (data set composition is described in the methods) and IRb (Inverted Repeat region). Support values are maximum likelihood bootstrap support/Bayesian posterior probability. The families of Myrtales are indicated by different colors. For each figure, the inset shows the same tree as a phylogram (except for some inconsistencies in the phylogenetic relationships of IR dataset construction). The support value on the branch is bootstrap value/Bayesian posterior probability: “*” means 100% /1.0 support value, and “-” means bootstrap value/Bayesian posterior probability is less than 60 / 0.7. The families of Myrtales are represented by different colors. The small picture in the upper left corner is the ML phylogenetic tree (showing branch length).
Additional file 2: Figure S7.
Optimal phylogenetic tree resulting from analyses of 92 complete chloroplast genomes of Myrtales and 20 outgroups using Maximum Likelihood (ML). Support values are maximum likelihood bootstrap support posterior probability. The families of Myrtales are indicated by different colors. . The support value on the branch is bootstrap value, “*” means 100% support value, and “-” means bootstrap value is less than 60. The families of Myrtales are represented by different colors. The small picture in the upper left corner is the ML phylogenetic tree (showing branch length).
Additional file 3: Table S1.
Eta, Pi value, H, Hd, PICs, the length and aligned length of 188 Myrtales homologous loci across.
Additional file 4: Table S2.
Species information and chloroplast genomes GenBank accession number of Outgroups in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, XF., Landis, J.B., Wang, HX. et al. Comparative analysis of chloroplast genome structure and molecular dating in Myrtales. BMC Plant Biol 21, 219 (2021). https://doi.org/10.1186/s12870-021-02985-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-021-02985-9