
Abstract
Background
Taxol is an efficient anticancer drug; however, the accumulation of taxoids can vary hugely among Taxus species. The mechanism underlying differential accumulation of taxoids is largely unknown. Thus, comparative analysis of the transcriptomes in three Taxus species, including T. media, T. mairei and T. cuspidata, was performed.
Results
KEGG enrichment analysis revealed that the diterpenoid biosynthesis and cytochrome P450 pathways were significantly enriched in different comparisons. Differential expressions of these taxol biosynthesis related genes might be a potential explanation for the interspecific differential accumulation of taxol and its derivatives. Besides, the sequences of several MEP pathway-associated genes, such as DXS, DXR, MCT, CMK, MDS, HDS, HDR, IPPI, and GGPPS, were re-assembled based on independent transcriptomes from the three Taxus species. Phylogenetic analysis of these MEP pathway-associated enzymes also showed a high sequence similarity between T. media and T. cuspidata. Moreover, 48 JA-related transcription factor (TF) genes, including 10 MYBs, 5 ERFs, 4 RAPs, 3 VTCs, and 26 other TFs, were analyzed. Differential expression of these JA-related TF genes suggested distinct responses to exogenous JA applications in the three Taxus species.
Conclusions
Our results provide insights into the expression pattern and sequence similarity of several taxol biosynthesis-related genes in three Taxus species. The data give us an opportunity to reveal the mechanism underlying the variations in the taxoid contents and to select the highest-yielding Taxus species.
Similar content being viewed by others

Background
Taxol (generic name paclitaxel), very important for certain types of cancer treatments, was first isolated from the bark of the Pacific yew Taxus brevifolia and gained marketing approval from the U.S. Food and Drug Administration for the treatment of various cancers [1]. Limited by several barriers, such as the slow growth of wild yews, destructive harvesting techniques, and complicated purification procedures, the demand for taxol exceeds the supply [2].
The metabolic pathway of taxol biosynthesis has been partially revealed. Firstly, three units of the C5 isoprenoid precursor isopentenyl diphosphate (IPP) and one unit of dimethylallyl diphosphate, which are produced by the plastidial 2-C-methyl-D-erythritol phosphate (MEP) pathway, are used to synthesize geranylgeranyl diphosphate (GGPP), the precursor of the diterpenoid taxane core [3]. The cyclization of GGPP to taxa-4(5),11(12)-diene is conducted by taxadiene synthase, which catalyzes a slow, but not rate-limiting step [4, 5]. Then, the pathway toward taxol involves many intermediate enzymatic steps, including eight hydroxylations, five acyl/aroyl transfer reactions, one epoxidation, one oxidation, two CoA esterifications and one N-benzoylation [6]. For example, several cytochrome P450s, including 2α-, 5α-, 7β-, 9α-, 10β-, 13α- and 14β-hydroxylases, participate in the oxygenation steps of taxadiene [7]. The assembly of the C13-side chain appended to baccatin III is considered to be the final step of the taxol biosynthesis pathway [8].
RNA-seq has been frequently used to evaluate the expression differences among different Taxus species. The first transcriptome of the Taxus genus was obtained from the cultured cells of T. cuspidata in 2010, and then the organ-specific transcriptomes of T. mairei were published in the same and next year [9, 10]. The deep sequencing of different Taxus species indicated roles of methyl-jasmonate (MeJA) in the regulation of the terpenoid biosynthesis pathway, which supplies the precursors for taxol biosynthesis [11,12,13]. A comparison between T. media and T. mairei transcriptomes provided insights into the differential expressions of candidate genes involved in the taxoid biosynthetic pathways [14]. Transcriptome profiles of T. chinensis under different temperatures revealed an involvement of various TF families, such as NAC, WRKY, bZIP, MYB, and ERF families, in the responses of Chinese yews to cold stress [15]. Recently, transcriptome assembly and systematic identification of the cytochrome P450 and WRKY families in T. chinensis have been performed by Yu’s group [16, 17]. In a novel T. yunnanensis cultivar, transcriptome profiling illustrated a specific regulation mechanism of taxol biosynthesis [18].
Recent technical advances in the large-scale identification of genes have revealed several complex processes involved in the regulation of plant metabolism [19, 20]. In nature, the amount of each taxoid varied among varieties and species [21,22,23]. Thus, investigations into the variations in the expression pattern and sequence similarity of the key genes involved in the metabolism of taxoids in three different Taxus species will provide an opportunity to select the highest-yielding species and to elucidate the mechanism underlying the species-related variations in the taxoid contents.
Methods
Plant materials and RNA extraction
Fresh twig samples were harvested from three-year-old cultivated Taxus trees, including T. media, T. mairei, and T. cuspidata, in March 2015 in a growth chamber of Hangzhou Normal University, Hangzhou, China (Fig. 1a). The growth conditions were set at 25 ± 1 °C with a light/dark cycle of 12/12 h and a 55–65% relative humidity. Five independent trees for each species were used in our study.

Variation of the contents of taxoids among three different Taxus species. a A picture of T. media, T. mairei and T. cuspidata under greenhouse condition. Fresh twigs were harvested from three cultivated Taxus species. b The contents of paclitaxel were quantified by HPLC-MS/MS method. A P value < 0.05 was considered to be statistically significant and indicated by “b” and P < 0.01 was indicated by “a”
For RNA isolation, we used the methodology previously described by Yu et al. [14]. In detail, total RNAs were extracted using an RNeasy plant mini kit (Qiagen, Hilden, Germany) according to its manual. DNA contamination was cleaned by adding DNase I to the mixture. The clean RNAs were checked using 1% agarose gel electrophoresis. The quality of total RNAs was confirmed using an RNA 6000 Nano LabChip kit (Agilent, Santa Clara, CA, USA) with an RNA integrity number > 7.0.
Library preparation and transcriptomic analysis
Total RNA samples of 10 μg from each RNA extract (3 species × 3 biological replicates) were prepared. The methods of library preparation, de novo strategy and transcriptomic analysis were the same to our previous published work [14]. In brief, RNA representing a specific mRNA was subjected to oligo (dT) attached magnetic beads. Then, purified RNAs were fragmented into small pieces. These small fragments were reverse-transcribed to create cDNA libraries using a sample preparation kit (Illumina, San Diego, USA). Sequencing was carried out using an Illumina Hiseq 4000 platform (LC-Bio, Hangzhou, China) according to its protocol.
Three important parameters, including Q20, Q30 and GC content, were verified to evaluate all the reads using FastQC online tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). For de novo assembly, the transcriptome from each species was processed using software Trinity 2.4.0 [24]. For gene identification and expression analysis, the reads from different species were co-assembled, and for gene sequence analysis, the reads from different species were assembled separately. The raw sequence data has been submitted to the NCBI Short Read Archive with accession number GSE121523.
Functional annotation and enrichment analysis
For gene annotation, we used the methodology previously described by Yu et al. [14]. In detail, all assembled unigenes were searched against various databases, including the non-redundant (Nr) protein, Gene Ontology (GO), SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), and eggNOG databases, with a threshold of E value < 0.00001. GO and KEGG enrichment analysis were performed on the DEGs by perl scripts in house.
Differentially expressed unigene (DEGs) analysis
Expression levels for each unigene were calculated using the TPM method [25]. The DEGs were screened with criterions: ǀlog2(fold change) ǀ > 1 and statistical significance P < 0.05. The transcript abundance values were transformed into Z-score after log transformation. The heatmap was drawn using MultiExperiment Viewer (version 4.9.0) basing on their log2 values of transformed Z-scores.
Homology analysis and phylogenetic tree building
Gene homology analysis was carried out using ClustalW with default parameters. Predicted full-length protein sequences of the key genes involved in the taxol biosynthesis pathway were used for alignments. The results were subsequently visualized by the GeneDoc software and a tree was constructed using software MEGA6.1 employing NJ method.
Analysis of paclitaxel contents
Paclitaxel (≥ 99%; CAS No. 33069–62-4) was purchased from Aladdin Biochemical Technology (Shanghai, China). Fresh twigs of each sample were collected, dried at 40 °C for 3 d, and powdered. A previously published method was used to prepare crude extracts and paclitaxel determination [14]. The quantification of paclitaxel was presented as the means of at least three replicates ± standard error.
Statistical analysis
For the transcriptome analyses, the false discovery rate was used to calculate the resulting P values, and the P values were adjusted using the Benjamini and Hochberg method. Statistical analyses were performed using SPSS software version 19.0 (SPSS Inc., Chicago, IL, USA), and an ANOVA was applied to compare the differences between two groups.
Results
Variations in taxol contents using a HPLC-MS/MS approach
To determine more precisely the differences in taxol contents among the three Taxus species, a HPLC-MS/MS approach was used. The data showed that T. cuspidata contained the highest level of taxol (1.67 mg.g− 1, DW) and T. mairei contained the lowest levels of taxol (0.66 mg.g− 1, DW) (Fig. 1b).
Transcriptomes of different Taxus species
The raw reads were qualified, and adapters were removed, yielding 67.49 Gb of sequence data, including 19.84 Gb from T. media, 21.90 Gb from T. cuspidata, and 25.75 Gb from T. mairei (Additional file 1). Pair-wise Pearson’s correlation coefficients of three replicates × three Taxus species indicated high repeatability of the sequencing data (Fig. 2a). To obtain an overview of the transcriptomic variations, a principal components analysis (PCA) was performed, and the explained values of PC1 and PC2 were 18.4 and 36.2%, respectively (Additional file 2). The PCA clearly separated the three species into two groups, suggesting a close similarity between T. media and T. cuspidata.

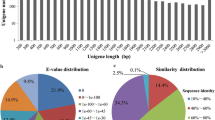
Illumina sequencing and transcriptomes of different Taxus species. a Pair-wise Pearson’s correlation coefficients of the sequencing data from three replicates × three Taxus species. The detail information of the assembled transcripts (b) and unigenes (c). d The size distributions of transcripts and unigenes of Taxus species. e The annotation of unigenes basing on various databases. f The species distribution of the annotated unigenes
All of the reads obtained from the three Taxus species were assembled, resulting in 52,261 transcripts (N50: 1472), with a mean length of 959 bp (Fig. 2b), and 33,895 unigenes (N50: 1373), with a mean length of 841 bp (Fig. 2c). The size distributions of transcripts and unigenes are shown in Fig. 2d. For transcripts, 10.98% of the reads were > 2000 bp in length, and the majority of the reads (64.06%) were < 1000 bp in length. Only 8.87% of the unigenes were > 2000 bp in length, and the majority of unigenes were between 200 bp and 500 bp in length. In total, 14,268 unigenes were annotated by the Swiss-Prot database, 22,074 unigenes were identified in the Nr database, 16,894 unigenes displayed significant similarities to known proteins in the Pfam database, and 8426 and 17,924 unigenes were annotated in the KEGG and KOG databases, respectively (Fig. 2e). The species distribution of the annotated unigenes is shown in Fig. 2f.
Identification of the DEGs among different Taxus species
In our study, a large number of unigenes were classified into various KEGG metabolic and signaling pathways (Additional file 3). The most enriched KEGG pathways were ‘amino acid metabolism’ (608 unigenes), ‘energy metabolism’ (638 unigenes), and ‘carbohydrate metabolism’ (796 unigenes) (Fig. 3a).

Identification of the DEGs among three different Taxus species. a Classification of enriched KEGG terms. A large number of unigenes could be classified into various KEGG metabolic and signaling pathways, including pathways related to organismal systems, metabolism, genetic information processing, environmental information processing, and cellular process. b A venn diagram showed the numbers of the DEGs in three comparisons, including T. media vs T. cuspidata, T. media vs T. mairei and T. mairei and T. cuspidata comparisons. c The numbers of the up- and down-regulated unigenes in the three comparisons. d KEGG enrichment analysis of the DEGs in the three comparisons. The significant P values of each KEGG term in the three comparisons were shown by a heatmap. The bar indicated the significant values. Red arrows indicated two metabolic pathways involved in the taxol biosynthesis pathway
The numbers of DEGs identified in each comparison are shown in a Venn diagram (Fig. 3b). In detail, 4726 T. media highly-expressed unigenes and 3030 T. cuspidata highly-expressed unigenes were identified in the T. media vs. T. cuspidata comparison. In the T. media vs. T. mairei comparison, 6750 unigenes highly expressed in T. media and 8095 unigenes highly expressed in T. mairei. In the T. mairei vs. T. cuspidata comparison, 6143 T. mairei highly-expressed unigenes and 9060 T. cuspidata highly-expressed unigenes were identified (Fig. 3c).
Among these pathways, 26 pathways were significantly enriched (P < 0.05) in the T. media vs. T. cuspidata comparison. In the T. media vs. T. mairei comparison, 18 pathways were significantly enriched, while in the T. mairei vs. T. cuspidata comparison, only 11 pathways were significantly enriched (Fig. 3d).
DEGs associated with the taxol biosynthesis pathway
For taxol biosynthesis, several intermediate steps, including precursor supply, diterpenoid taxane core synthesis, hydroxylations, acylations, baccatin III formation, and C13-side chain assembly, were involved (Fig. 4a) [6]. A transcriptomic analysis revealed six taxol biosynthesis-related GO terms, including ‘paclitaxel biosynthetic process’ (GO:0042617), ‘taxane 10-beta-hydroxylase activity’ (GO:0050597), ‘2-alpha-hydroxytaxane 2-O-benzoyltransferase activity’ (GO:0050642), ‘taxadiene 5-alpha-hydroxylase activity’ (GO:0050604), ‘taxane 13-alpha-hydroxylase activity’ (GO:0050598), and ‘taxadiene synthase activity’ (GO:0050553), and provided an opportunity to analyze the differential expression of taxol biosynthesis pathway-related genes among different Taxus species. In detail, the genes in four GO terms, including GO:0042617 (P = 1.05E-9), GO:0050597 (P = 3.21E-4), GO:0050642 (P = 1.11E-3) and GO:0050604 (P = 8.36E-3), significantly changed in the T. media vs. T. cuspidata comparison (Fig. 4b).

Differential expression of the unigenes related to the taxol biosynthesis pathway. a Overview of the taxol biosynthesis pathway. b Enrichment analysis of six taxol biosynthesis-related GO terms. The significant P values of each GO term in the three comparisons were shown by a heatmap. c Expression analysis of the unigenes related to the taxol biosynthesis pathway. Enzymes abbreviations are: DXS: 1-Deoxy-D-xylulose 5-phosphate synthase; DXR: 1-Deoxy-D-xylulose 5-phosphate reductoisomerase; MCT: 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CMK: 4-(Cytidine 5-diphospho)-2-C-methyl-D-erythritol kinase; MDS: 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; HDS: 4-Hydroxy-3-methylbut-2-enyl-diphosphate synthase; HDR: 4-Hydroxy-3-methylbut-2-enyl diphosphate reductase; IPPI: isopentenyl diphosphate isomerase; GGPPS: geranylgeranyl diphosphate synthase; TS, taxadiene synthase; T13OH: taxoid 13-α-hydroxylase; T10OH: taxoid 10-β-hydroxylase; T14OH: Taxoid 14-β-hydroxylase; T7OH: taxoid-7β-hydroxylase; T2OH: taxoid-2α-hydroxylase; T5OH: taxadiene-5α-hydroxylase; DBAT: 10-deacetylbaccatin III-10-β-O-acetyltransferase; PAL: phenylalanine aminomutase; Benzoyl CoA: 2-debenzoyl-7,13-diacetylbaccatin III-2-O-benzoyl transferase. The bar indicated the “log2(expression folds)”
Furthermore, the expression patterns of the taxol biosynthesis pathway-related genes were investigated. In the MEP pathway, most of the genes showed highest expression levels in T. media. For the diterpenoid taxane core biosynthesis, the gene (comp26263_c0) encoding the key enzyme, taxadiene synthase, predominantly expressed in T. cuspidata. For the hydroxylation and acylations steps, a series of taxane hydroxylase-encoding genes was identified, and most of them highly expressed in T. cuspidata. For the C13-side chain assembly, one phenylalanine ammonia-lyase encoding gene (com13676_c0) and two benzoyl-CoA encoding genes (comp28838_c0 and comp4855_c0) were identified, and they predominantly expressed in T. cuspidata (Fig. 4c).
Phylogenetic analysis of the MEP pathway-associated proteins
From the independently assembled transcriptomes of three Taxus species, the full-length sequences of 12 MEP pathway-related genes were obtained. Three predicted peptide sequences of each key enzyme were used for phylogenetic tree construction. For most of the MEP pathway-related enzymes, including 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), 1-deoxy-D-xylulose 5-phosphate synthase 1 (DXS1), DXS2, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MDS), 4-(cytidine 5-diphospho)-2-C-methyl-D-erythritol kinase (CMK), GPPS, geranylgeranyl diphosphate synthase 1 (GGPPS1), and GGPPS2, sequences from T. cuspidata were highly similar to those from T. media. The sequences of three MEP pathway-related enzymes, 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (HDR), 4-hydroxy-3-methylbut-2-enyl-diphosphate synthase (HDS), and isopentenyl diphosphate isomerase (IPPI), displayed high similarity levels between T. media and T. cuspidata (Additional file 4).
Transcriptomic analysis reveals the differences in jasmonic acid (JA) pathway-related genes
Based on the transcriptomes, four JA metabolism-related GO terms, ‘regulation of jasmonic acid mediated signaling pathway’ (GO:2000022), ‘response to jasmonic acid stimulus’ (GO:0009753), ‘jasmonic acid mediated signaling pathway’ (GO:0009867), and ‘cellular response to jasmonic acid stimulus’ (GO:0071395), were identified. In detail, the significant values of GO:2000022 in the T. mairei vs. T. cuspidata and T. media vs. T. mairei comparisons were 0.044 and 0.034, respectively. For GO:0009753, the significant values in the T. mairei vs. T. cuspidata and T. media vs. T. mairei comparisons were 0.048 and 0.033, respectively (Additional file 5). There were no significant differences in these terms in the T. media vs. T. cuspidata comparison. Significant differences for GO:2000022 and GO:0009753 were observed in the T. media vs. T. mairei and T. mairei vs. T. cuspidata comparisons (Fig. 5a). The expression pattern of 48 JA metabolism- and signaling pathway-related genes was analyzed (Fig. 5b). Interestingly, the expression pattern of these JA-related genes in T. media was similar to that in T. cuspidata (Additional file 6). Furthermore, the endogenous JA contents were determined in the three Taxus species. Significant differences in the endogenous JA content were observed in T. media vs. T. mairei and T. mairei vs. T. cuspidata comparisons (Fig. 5c).

Analysis of transcriptome reveals the differences in JA pathway-related genes. a Significance analysis of five JA-related GO terms in three comparisons, including T. media vs T. cuspidata, T. media vs T. mairei and T. mairei and T. cuspidate comparisons, were shown by a heatmap. b Expression profiles of the transcription factor encoding genes related to JA signaling pathway were showed by a heatmap. c Determination of endogenous JA contents in three Taxus species. A P value > 0.05 was considered to be statistically significant and indicated by “b” and P < 0.05 was indicated by “a”
Identifications of TFs families in Taxus species
A number of TFs were reported to play important roles in taxol biosynthesis. In our study, 736 putative TF encoding genes belonging to 17 major TF families were analyzed in Taxus (Additional file 7). A large number of TFs were included in the MYB family (174 genes), ARF family (90 genes), and WRKY family (61 genes). The numbers of differential expressed TFs in the three comparisons were showed in Additional file 8. To screen key regulators for taxol biosynthesis, the expression levels of all TF genes were showed in a heatmap (Additional file 9).
Discussion
Because Taxus plants are the major natural resource for taxol, comprehensive phytochemical analyses of Taxus species have been performed [6, 26]. A large number of compounds have been identified in various Taxus species [27]. However, the levels of taxoids accumulation may vary significantly among species. The previous study showed that the taxol contents in T. mairei, T. media, and T. cuspidata needles were 0.163 mg.g− 1, 0.435 mg.g− 1, and 0.249 mg.g− 1, respectively, by UFLC-ESI-MS and UFLC-DAD analysis [28]. In our study, T. mairei contained the lowest levels of taxol, which was in agree with the results from the previous work. However, the contents of taxol in T. cuspidata was higher than that in T. media. Another previous study showed that the taxol contents ranged from 0.1 to 0.4 mg.g− 1 in T. mairei needles in the course of a year [29]. Rikhari’s group showed that taxol content in the bark of Himalayan yew was related to tree age and sex [30]. The accumulation of taxol was greatly affected by natural environment and cultivation conditions, thus the taxol contents varied among different studies.
A substantial number of DEGs, among which 26 major metabolic pathways were enriched, were identified in the T. media vs. T. cuspidata, T. media vs. T. mairei, and T. mairei vs. T. cuspidata comparisons (Fig. 3b, c). Formation of diterpenoid taxane skeleton is an essential step for taxol biosynthesis, and it occurs before the cyclization of taxane skeleton [4]. In our study, the diterpenoid biosynthesis pathway significantly changed in the T. media vs. T. cuspidata and T. media vs. T. mairei comparisons. No significant changes were observed in the T. mairei vs. T. cuspidata comparison (Fig. 3d). It suggested that the genes involved in diterpenoid biosynthesis shared a similar expression pattern between T. mairei and T. cuspidata. Additionally, the drug metabolism–cytochrome P450 pathway, containing a series of taxoid oxygenases, was significantly different in all three comparisons [7, 31]. The differential expressions of the above two important metabolic pathways might provide a number of genes that were involved in the interspecific differential accumulations of taxol and its derivatives. In the taxol biosynthetic pathway, α- and β-phenylalanine are involved in the side chain assembly [32, 33]. KEGG analysis showed that the phenylalanine metabolism pathway was siginificantly enriched in all three comparisons, suggesting an involvement of side chain assembly in interspecific differential accumulations of taxol. Additionally, flavonoids and phenylpropanoids are important active ingredients isolated from Taxus reminder extracts [23, 34, 35]. Two enriched flavonoid metabolism-related pathways, including the flavonoid biosynthesis and flavone and flavonol biosynthesis pathways, were identified in the T. media vs T. cuspidata comparison. Our data indicated that there were great differences in the accumulation of flavonoids between T. media and T. cuspidata. The phenylpropanoid biosynthesis pathway was enriched in all three comparisons, suggesting deep variations in various active ingredient metabolisms, not only taxol, among different Taxus species.
Previous studies identified a number of taxol biosynthesis-related genes that were assigned into six GO terms [12, 36]. In the T. baccata plantlets, the taxane contents were correlated with the expression levels of TXS, DBAT, BAPT, and DBTNBT genes [37]. In our study, there was a great difference in the expression of taxol biosynthesis-related genes between T. media and T. cuspidata (Fig. 4b). The expression pattern of the taxol biosynthesis-related genes, including precursor supply (10 genes), diterpenoid taxane core (4 genes), hydroxylation and acylations (14 genes), baccatin III formation (2 genes), and C13-side chain assembly (3 genes), was investigated. The dynamic expression levels of these genes might provide a potential explanation for the interspecific differential accumulation of taxol.
The sequences and structural properties of key enzymes in the taxol biosynthesis pathway, which result in different catalytic efficiencies, are closely related to taxol production [38, 39]. For example, improvement of the 10-deacetylbaccatin III-10-β-O-acetyltransferase (DBAT) catalytic fitness contributes to the abundant accumulation of baccatin III [40]. Sequences of several key enzymes were re-assembled based on the independent transcriptomes from each Taxus species. Due to the unavailability of corresponding genome data, the assembled sequences of most taxol pathway-related genes were imperfect. Fortunately, the full-length sequences of most MEP pathway genes were available. Interestingly, for most MEP pathway genes, such as DXR, DXS, MDS, CMK, GPPS, and GGPPS, greater sequence similarities were observed between T. cuspidata and T. media. This result was consistent with the differential accumulation of taxol among the three Taxus species.
JA and MeJA have been widely applied to increase secondary metabolite production in various culture systems [41]. Exogenous MeJA has also been used as an effective elicitor to increase the production of paclitaxel and its derivatives in Taxus cell-suspension cultures [11, 42]. Considering the highest accumulation of endogenous JA in T. mairei, the level of endogenous JAs may not reach the threshold to play a role in taxol biosynthesis. Several TFs were involved in the JA-mediated transcriptional regulation of secondary metabolism in medicinal plants [43]. In our study, a number of JA pathway-related genes were identified, and their expression levels were also analyzed in the three Taxus species. Among these genes, some TFs, such as MYB, LHY, and ERF, were included (Fig. 5b). The differential expression of these JA-related TFs suggested distinct responses of the three Taxus species to exogenous MeJA application.
Furthermore, increasing evidences showed that various TF families, such as bHLH and WRKY, involved in the regulation of taxol biosynthesis [32]. For example, TcMYC1, TcMYC2, and TcMYC4 are involved in transduction of JA signals and regulation of taxol biosynthesis [44, 45]. A WRKY TF, TcWRKY1, plays a role in transcriptional activation of DBAT in T. chinensis [46]. In our study, 43 bHLH family and 61 WRKY family TFs were identified. More than half of these bHLH TFs and most of these WRKY TFs were identified as DEGs in the T. media vs. T. cuspidata and T. media vs. T. mairei comparisons, providing a number of candidate regulators of taxol biosynthesis.
Conclusions
Differential expressions of the genes involved in taxol biosynthesis pathway, including precursor supply-, diterpenoid taxane core-, hydroxylation and acylation-, baccatin III formation-, and C13-side chain assembly-related genes, might provide a potential explanation for the interspecific differential accumulation of taxol in different Taxus species. Phylogenetic analysis indicated that sequence diversity in the MEP pathway genes may be another factor that determines the variations in taxoids. Our results contribute to a deeper understanding of the interspecific differential accumulation of taxoids in three Taxus species.
Abbreviations
- 10-DAB:
-
10-deacetylbaccatin-III
- ANOVA:
-
Analysis of variance
- CMK:
-
4-(Cytidine 5-diphospho)-2-C-methyl-D-erythritol kinase
- DEG:
-
Differential expressed genes
- DXR:
-
1-Deoxy-D-xylulose 5-phosphate reductoisomerase
- DXS1:
-
1-Deoxy-D-xylulose 5-phosphate synthase 1
- GGPP:
-
Geranylgeranyl diphosphate
- GO:
-
Gene Ontology
- JA:
-
Jasmonic acid;
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MDS:
-
2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase
- MeJA:
-
Methyl-jasmonate
- MEP:
-
2-C-methyl-D-erythritol phosphate
- UPLC-MS/MS:
-
Ultra Performance Liquid Chromatography-Coupled Mass Spectrometry
References
Wani MC, Horwitz SB. Nature as a remarkable chemist: a personal story of the discovery and development of Taxol. Anti-Cancer Drugs. 2014;25(5):482–7.
Goodman J, Walsh V. The story of taxol: nature and politics in the pursuit of an anticancer drug. New York: Cambridge University Press. 2001;107(2):577-8.
Eisenreich W, Menhard B, Hylands PJ, Zenk MH, Bacher A. Studies on the biosynthesis of taxol: the taxane carbon skeleton is not of mevalonoid origin. Proc Natl Acad Sci U S A. 1996;93(13):6431–6.
Harrison JW, Scrowston RM, Lythgoe B, Harrison JW, Scrowston RM, Lythgoe B. Taxine. Part IV. The constitution of taxine-I. J Chem Soc C Organic. 1966:1933–45.
Williams DC, Wildung MR, Jin AQ, Dalal D, Oliver JS, Coates RM, et al. Heterologous expression and characterization of a “Pseudomature” form of taxadiene synthase involved in paclitaxel (Taxol) biosynthesis and evaluation of a potentialintermediate and inhibitors of the multistep diterpene cyclization reaction. Arch Biochem Biophys. 2000;379(1):137–46.
Croteau R, Ketchum RE, Long RM, Kaspera R, Wildung MR. Taxol biosynthesis and molecular genetics. Phytochem Rev. 2006;5(1):75–97.
Kaspera R, Croteau R. Cytochrome P450 oxygenases of Taxol biosynthesis. Phytochem Rev. 2006;5(2–3):433–44.
Jiménez-Barbero J, Amat-Guerri F, Snyder JP. The solid state, solution and tubulin-bound conformations of agents that promote microtubule stabilization. Curr Med Chem Anticancer Agents. 2002;2(1):91–122.
Hao DC, Ge G, Xiao P, Zhang Y, Yang L. The first insight into the tissue specific Taxus transcriptome via illumina second generation sequencing. PLoS One. 2011;6(6):e21220.
Lee E-K, Jin Y-W, Park JH, Yoo YM, Hong SM, Amir R, et al. Cultured cambial meristematic cells as a source of plant natural products. Nat Biotech. 2010;28(11):1213–7.
Li ST, Zhang P, Zhang M, Fu CH, Zhao CF, Dong YS, et al. Transcriptional profile of Taxus chinensis cells in response to methyl jasmonate. BMC Genomics. 2012;13:295.
Sun G, Yang Y, Xie F, Wen JF, Wu J, Wilson IW, et al. Deep sequencing reveals transcriptome re-programming of Taxus × media cells to the elicitation with methyl jasmonate. PLoS One. 2013;8(4):e62865.
Patil RA, Lenka SK, Normanly J, Walker EL, Roberts SC. Methyl jasmonate represses growth and affects cell cycle progression in cultured Taxus cells. Plant Cell Rep. 2014;33(9):1479–92.
Yu C, Guo H, Zhang Y, Song Y, Pi E, Yu C, et al. Identification of potential genes that contributed to the variation in the taxoid contents between two Taxus species (Taxus media and Taxus mairei). Tree Physiol. 2017;37(12):1659–71.
Meng D, Yu X, Ma L, Hu J, Liang Y, Liu X, et al. Transcriptomic response of Chinese yew (Taxus chinensis) to cold stress. Front Plant Sci. 2017;8:468.
Zhang M, Chen Y, Nie L, Jin X, Liao W, Zhao S, et al. Transcriptome-wide identification and screening of WRKY factors involved in the regulation of taxol biosynthesis in Taxus chinensis. Sci Rep. 2018;8(1):5197.
Liao W, Zhao S, Zhang M, Dong K, Chen Y, Fu C, et al. Transcriptome assembly and systematic identification of novel cytochrome P450s in Taxus chinensis. Front Plant Sci. 2017;8:1468.
He CT, Li ZL, Zhou Q, Shen C, Huang YY, Mubeen S, et al. Transcriptome profiling reveals specific patterns of paclitaxel synthesis in a new Taxus yunnanensis cultivar. Plant Physiol Biochem. 2018;122:10–8.
Jin J, Zhang H, Zhang J, Liu P, Chen X, Li Z, et al. Integrated transcriptomics and metabolomics analysis to characterize cold stress responses in Nicotiana tabacum. BMC Genomics. 2017;18(1):496.
Xu Q, Zhu J, Zhao S, Hou Y, Li F, Tai Y, et al. Transcriptome profiling using single-molecule direct RNA sequencing approach for in-depth understanding of genes in secondary metabolism pathways of Camellia sinensis. Front Plant Sci. 2017;8:1205.
Poupat C, Hook I, Gueritte F, Ahond A, Guenard D, Adeline MT, et al. Neutral and basic taxoid contents in the needles of Taxus species. Planta Med. 2000;66(6):580–4.
Yu C, Luo X, Zhan X, Hao J, Zhang L, YB LS, et al. Comparative metabolomics reveals the metabolic variations between two endangered Taxus species (T. fuana and T. yunnanensis) in the Himalayas. BMC Plant Biol. 2018;18(1):197.
Hao J, Guo H, Shi X, Wang Y, Wan Q, Song Y, et al. Comparative proteomic analyses of two Taxus species (Taxus × media and Taxus mairei) reveals variations in the metabolisms associated with paclitaxel and other metabolites. Plant Cell Physiol. 2017;114(24):6388.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621.
Zheng W, Komatsu S, Zhu W, Zhang L, Li X, Cui L, et al. Response and defense mechanisms of Taxus chinensis leaves under UV-A radiation are revealed using comparative proteomics and metabolomics analyses. Plant Cell Physiol. 2016;57(9):1839–53.
Li C, Huo C, Zhang M, Shi Q. Chemistry of Chinese yew, Taxus chinensis var. mairei. Biochemical Syst Ecol. 2008;36(4):266–82.
Ge GB, Liang SC, Hu Y, Liu XB, Mao YX, Zhang YY, et al. Rapid qualitative and quantitative determination of seven valuable taxanes from various Taxus species by UFLC-ESI-MS and UFLC-DAD. Planta Med. 2010;76(15):1773–7.
Yang L, Zheng ZS, Cheng F, Ruan X, Jiang DA, Pan CD, et al. Seasonal dynamics of metabolites in needles of Taxus wallichiana var. mairei. Molecules. 2016;21(10).
Nadeem M, Rikhari HC, Kumar A, Palni LM, Nandi SK. Taxol content in the bark of Himalayan yew in relation to tree age and sex. Phytochemistry. 2002;60(6):627–31.
Rasool S, Mohamed R. Plant cytochrome P450s: nomenclature and involvement in natural product biosynthesis. Protoplasma. 2016;253(5):1197–209.
Yanfang Y, Kaikai Z, Liying Y, Xing L, Ying W, Hongwei L, et al. Identification and characterization of MYC transcription factors in Taxus sp. Gene. 2018;675:1–8.
Klettke KL, Sanjit S, Washington M, Walker KD. Beta-styryl- and beta-aryl-beta-alanine products of phenylalanine aminomutase catalysis. J Am Chem Soc. 2007;129(22):6988.
Ruan X, Zhan LM, Gao XX, Yan LY, Zhang H, Zhu ZY, et al. Separation and purification of flavonoid from Taxus remainder extracts free of taxoids using polystyrene and polyamide resin. J Sep Sci. 2013;36(12):1925–34.
Jin-Kyu K, Yeon-Ho H, Karchesy JJ, Young-Soo B. A new phenylpropanoid derivative of catechin from the needles of Taxus cuspidata. Nat Prod Res. 2008;22(15):1365–9.
Nasiri J, Naghavi MR, Alizadeh H, Moghadam MR. Seasonal-based temporal changes fluctuate expression patterns of TXS, DBAT, BAPT and DBTNBT genes alongside production of associated taxanes in Taxus baccata. Plant Cell Rep. 2016;35(5):1103–19.
Onrubia M, Moyano E, Bonfill M, Palazon J, Goossens A, Cusido RM. The relationship between TXS, DBAT, BAPT and DBTNBT gene expression and taxane production during the development of Taxus baccata plantlets. Plant Sci. 2011;181(3):282–7.
Walker K, Croteau R. Taxol biosynthesis: molecular cloning of a benzoyl- CoA:taxane 2α-O-benzoyltransferase cDNA from Taxus and functional expression in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97(25):13591–6.
Dou T-Y, Luan H-W, Ge G-B, Dong M-M, Zou H-F, He Y-Q, et al. Functional and structural properties of a novel cellulosome-like multienzyme complex: efficient glycoside hydrolysis of water-insoluble 7-xylosyl-10-deacetylpaclitaxel. Sci Rep. 2015;5:13768.
Li B-J, Wang H, Gong T, Chen J-J, Chen T-J, Yang J-L, et al. Improving 10-deacetylbaccatin III-10-β-O-acetyltransferase catalytic fitness for Taxol production. Nat Commun. 2017;8:15544.
Pauwels L, Morreel K, De Witte E, Lammertyn F, Van Montagu M, Boerjan W, et al. Mapping methyl jasmonate-mediated transcriptional reprogramming of metabolism and cell cycle progression in cultured Arabidopsis cells. Proc Natl Acad Sci U S A. 2008;105(4):1380–5.
Yukimune Y, Tabata H, Higashi Y, Hara Y. Methyl jasmonate-induced overproduction of paclitaxel and baccatin III in Taxus cell suspension cultures. Nat Biotechnol. 1996;14(9):1129–32.
Sadia A, Jing-Jia H, Lou Z-Y. JA-mediated transcriptional regulation of secondary metabolism in medicinal plants. Sci Bull. 2015;60(12):1062–72.
Zhang M, Jin X, Chen Y, Wei M, Liao W, Zhao S, et al. TcMYC2a, a basic helix-loop-helix transcription factor, transduces JA-signals and regulates Taxol biosynthesis in Taxus chinensis. Front Plant Sci. 2018;9:863.
Lenka SK, Nims NE, Vongpaseuth K, Boshar RA, Roberts SC, Walker EL. Jasmonate-responsive expression of paclitaxel biosynthesis genes in Taxus cuspidata cultured cells is negatively regulated by the bHLH transcription factors TcJAMYC1, TcJAMYC2, and TcJAMYC4. Front Plant Sci. 2015;6:115.
Acknowledgements
We are also grateful to LC Sciences company (Hangzhou, China) for technical support. We are grateful to Kaidong Liu (Life Science and Technology School, Lingnan Normal University) for reading this manuscript.
Funding
Our work was funded by the Zhejiang Provincial Natural Science Foundation of China under Grant No. LY19C160001 and LY19C200010, National Key Research and Development Project (2016YFC0503100), and National Natural Science Foundation of China (31501810). The funding body played no role in our study.
Availability of data and materials
The datasets generated and analysed during the current study are available in the NCBI Short Read Archive with accession number GSE121523.
Author information
Authors and Affiliations
Contributions
CS and MD made substantial contributions to conception, was involved in drafting the manuscript, and given final approval of the version to be published. TZ, CY, XL, and YB made substantial contributions to acquisition of data analysis, was involved in revising the manuscript, and given final approval of the version to be published. LZ and CZ made substantial contributions to analysis and interpretation of data, was involved in revising the manuscript, and given final approval of the version to be published. All authors have read and approved the manuscript, and ensure that this is the case.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This project uses plant materials and does not utilize transgenic technology. The authorities responsible for the Taxus resources are the Mount Changbai National Nature Reserve, who provided permission to collect the samples of T. cuspidata and the Mount Tianmu National Nature Reserve, who provided permission to collect the samples of T. mairei, for our scientific research. T. media is cultivated variety purchased from the Wuwangnong company (Chengdu, China), who provided permission to use the seedlings for our scientific research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The detail information of raw reads from different sample groups. (XLSX 9 kb)
Additional file 2:
Figure S1. Principal components analysis of the three transcriptomes. (DOCX 14 kb)
Additional file 3:
Table S2. KEGG classification of the unigenes. (XLSX 580 kb)
Additional file 4:
Figure S2. Phylogenetic analysis of the proteins associated with the MEP pathway. (DOCX 412 kb)
Additional file 5:
Table S3. The significant values of each JA-related GO terms in various comparisons. (XLSX 9 kb)
Additional file 6:
Table S4. The expression levels of the JA-related genes in different Taxus species. (XLSX 17 kb)
Additional file 7:
Table S5. Identifications of transcription factors of the three Taxus species. (XLSX 205 kb)
Additional file 8:
Table S6. The numbers of differential expressed TFs in the three comparisons. (XLSX 9 kb)
Additional file 9:
Figure S3. A heatmap of differential expressed TF genes in the three comparisons. (DOCX 283 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, T., Luo, X., Yu, C. et al. Transcriptome analyses provide insights into the expression pattern and sequence similarity of several taxol biosynthesis-related genes in three Taxus species. BMC Plant Biol 19, 33 (2019). https://doi.org/10.1186/s12870-019-1645-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1645-x