
Abstract
Background
Grain length, as a critical trait for rice grain size and shape, has a great effect on grain yield and appearance quality. Although several grain size/shape genes have been cloned, the genetic interaction among these genes and the molecular mechanisms of grain size/shape architecture have not yet to be explored.
Results
To investigate the genetic interaction between two major grain length loci of rice, GS3 and qGL3, we developed two near-isogenic lines (NILs), NIL-GS3 (GS3/qGL3) and NIL-qgl3 (gs3/qgl3), in the genetic background of 93–11 (gs3/qGL3) by conventional backcrossing and marker-assisted selection (MAS). Another NIL-GS3/qgl3 (GS3/qgl3) was developed by crossing NIL-GS3 with NIL-qgl3 and using MAS. By comparing the grain lengths of 93–11, NIL-GS3, NIL-qgl3 and NIL-GS3/qgl3, we investigated the effects of GS3, qGL3 and GS3 × qGL3 interaction on grain length based on two-way ANOVA. We found that GS3 and qGL3 had additive effects on rice grain length regulation. Comparative analysis of primary panicle transcriptomes in the four NILs revealed that the genes affected by GS3 and qGL3 partially overlapped, and both loci might be involved in brassinosteroid signaling.
Conclusion
Our data provide new information to better understand the rice grain length regulation mechanism and help rice breeders improve rice yield and appearance quality by molecular design breeding.
Similar content being viewed by others


Background
When breeding cereal crops, the choice of a larger grain can increase the yield of crop varieties when other yield-related traits remain relatively stable. Among the three key components of rice yield (grain weight, panicles per plant and grain number per panicle), grain weight has high heritability [1]. Rice grains display a comparatively geometric shape, which can be broken down into grain length (GL), grain width (GW) and grain thickness (GT). These size/shape traits combined with grain density can explain the rice grain weight trait effectively.
Through linkage and association mapping, many quantitative trait loci (QTLs) for grain size/shape have been identified in different mutants or natural populations [2]. Only a small portion of these loci have been cloned, including GS3 [3–5], GL3.1/qGL3 [6, 7] and TGW6 [8] for grain length, and GW2 [9], GW5/qSW5 [10, 11], GS5 [12] and GW8 [13] for grain width. Some grain size/shape QTLs, such as gw8.1 [14], GW6 [15], qGL7 [16], qGL7-2 [17], GS7 [18] and qSS7 [19], were also mapped to a narrow chromosome region. Additionally, several small (or short) seed phenotype causal genes were identified by map-based cloning, including D1 [20–22], BU1 [23], SRS1 [24], SRS3 [25], SRS5 [26], and SG1 [27].
There are few reports about the genetic interaction of these characterized genes [2]. Yan et al. (2011) found genetic interactions between GS3 and qSW5. The effect of qSW5 on seed length was masked by GS3 alleles, and the effect of GS3 on seed width was masked by qSW5 alleles. No significant QTL interaction was observed between the two major grain width genes, GW2 and qSW5/GW5, suggesting that they might work to regulate grain width in independent pathways [28]. GS7 was effective in the presence of the GS3 non-functional A-allele and ineffective when combined with the functional GS3 C-allele [18]. However, how these genes work together or interact with others has not been deeply explored. The genetic interaction between two major grain length QTLs, GS3 and qGL3, also remains unclear. At least four different alleles for GS3 were identified by Mao et al. (2010): GS3-1 (Zhenshan 97), GS3-2 (Nipponbare), GS3-3/gs3 (Minghui 63) and GS3-4 (Chuan 7). GS3-1 and GS3-2 are functional short grain alleles, and GS3-4 is a stronger functional extra-short grain forming allele. GS3-3 has a premature termination, resulting in a non-functional long grain allele. At the cellular level, GS3 controls grain size largely by modulating the longitudinal cell number in grain glumes. Its organ size regulation domain in the N-terminus is necessary and sufficient for it to function as a negative regulator and act as a dominant allele [3]. One of its homologs in the rice genome, DENSE AND ERECT PANICLE1, also functions as a negative regulator of rice grain length [29, 30]. Recently, its homolog in Arabidopsis, AGG3, was shown to be an atypical heterotrimeric GTP-binding protein (G-protein) γ-subunit that positively regulated organ size [31, 32]. Another major grain-length locus, GL3.1/qGL3, was map-based cloned and characterized by two independent groups [6, 7]. GL3.1/qGL3 encodes a putative protein phosphatase (OsPPKL1) containing two Kelch domains. Transgenic studies showed that the Kelch domains functioned as a negative regulator and were essential for the biological function of OsPPKL1. At the cellular level, qGL3 functions by negatively modulating the longitudinal cell number in grain glumes.
In this study, we focused on the genetic interaction between two major grain length QTLs, GS3 and qGL3. The functional and non-functional alleles of GS3 and qGL3 were individually or simultaneously placed in the genetic background of 93–11 (an indica rice cultivar) to evaluate their genetic interaction. To understand these interactions at the molecular level, we analyzed the transcriptomes of young panicles (3–6 cm, glume development stage) of the NILs combining different alleles of GS3 and qGL3 through microarray assays. Our work could be helpful to better understand the genetic and molecular mechanisms of grain length regulation and molecular design rice breeding.
Results
The additive effects of GS3 and qGL3 on grain length
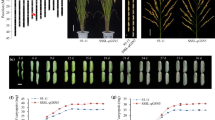
Functional GS3 and non-functional qgl3 were introduced into the 93–11 genetic background (genotype gs3/qGL3) to generate NIL-GS3 (genotype GS3/qGL3) and NIL-qgl3 (genotype gs3/qgl3), respectively. By crossing NIL-GS3 with NIL-qgl3, and marker-assisted selection (MAS), we created a third line, NIL-GS3/qgl3 (genotype GS3/qgl3). The grain lengths of these three NILs and their recurrent parent 93–11 with different allele combinations of GS3 and qGL3 were analyzed (Fig. 1a). We applied a two-way analysis of variance (ANOVA) for grain length (four NILs) and genotype (GS3 and qGL3), and observed significant additive effects on grain length for GS3 × qGL3 (P = 1.27 × 10−8), qGL3 (P = 3.71 × 10−13), and GS3 (P = 4.4 × 10−15) (Table 1). Considering NIL-GS3 (GS3/qGL3) as the control background, the loss of GS3 increased the grain length from 8.5 mm (GS3/qGL3) to 10.2 mm (gs3/qGL3), the loss of qGL3 increased the grain length from 8.5 mm (GS3/qGL3) to 11.2 mm (GS3/qgl3), and the loss of both increased the grain length from 8.5 mm (GS3/qGL3) to 12.2 mm (gs3/qgl3). Loss of qGL3 increased grain length more in the functional GS3 background (~2.7 mm) than in the non-functional gs3 background (~2.0 mm). Similarly, loss of GS3 increased grain length more in the functional qGL3 background (~1.7 mm) than in the non-functional qgl3 background (~1.0 mm) (Table 2). According to these data, we concluded that GS3 and qGL3 had additive effects larger than genetic interaction on rice grain length regulation and that the effects of qGL3 were stronger (Table 1).

Grains and plants of the NILs and comparison of their expression profiles. a Grains of the three NILs and their genetic background, 93–11. Scale bar, 10.0 mm. b Plants of three NILs and their genetic background, 93–11. Scale bar, 20.0 cm. c Venn diagram of the genes from different comparisons; red numbers indicate up-regulation, black indicates down-regulation. d Expression profiles of the genes commonly regulated by the comparisons gs3/qGL3 vs. GS3/qGL3, GS3/qgl3 vs. GS3/qGL3 and gs3/qgl3 vs. GS3/qGL3
The genetic interactions between GS3 and qGL3 on the expression levels of commonly regulated genes
Based on the microarray data, by comparing the differentially expressed genes in gs3/qGL3 vs. GS3/qGL3, GS3/qgl3 vs. GS3/qGL3, and gs3/qgl3 vs. GS3/qGL3, we found that seven genes were commonly up-regulated by > 1.5-fold (Fig. 1C, D and Table 3) and 37 genes were down-regulated by < 0.67-fold (Fig. 1c, d). Using gene expression levels (in 93–11 and its three NILs) and genotype (GS3 and qGL3) as the main factors, we applied a two-way ANOVA to the datasets from all four microarrays to identify the seven up-regulated genes significantly affected by GS3 and qGL3 (Table 3). There were significant GS3 × qGL3 interactions for the expression levels of the seven up-regulated genes with P-values < 0.05, except for Os03g40400 and Os04g59000 (Table 3). Based on two-way ANOVA analysis, we found a significant genetic interaction between GS3 and qGL3 according to the expression levels of the genes down-regulated by GS3 and qGL3 (Additional file 1: Table S7). Interestingly, the effects of GS3 and qGL3 on grain length was additive, on the expression levels of the commonly regulated genes it showed significant genetic interaction.
Among the seven genes up-regulated (>1.5-fold) by both gs3 and qgl3 (Fig. 1d), we found some encoded receptor protein kinases that might operate in the same signaling pathways to increase grain length in rice and explain the additive effects of gs3 and qgl3. Another commonly up-regulated gene, Os11g44880, was found to encode a kinesin-4, whose homolog, SRS3 (kinesin-13), was reported to positively regulate rice grain length [25]. Among the genes commonly down-regulated by gs3 and qgl3 (Fig. 1d), we found that gs3 and qgl3 down-regulated a gene (Os07g43670) encoding a ribonuclease T2 family domain-containing protein by 46- and 34-fold, respectively.
Profiling of gene up- and down-regulation and gene ontology analysis of DEGs in different genotypes
To reveal the genes affected by gs3 and qgl3, we compared the transcriptomes of the primary panicles of 93–11 (gs3/qGL3) and its three NILs through microarray analysis. Compared with the NIL-GS3 (GS3/qGL3) background, 92 genes were up-regulated by > 1.5-fold and 546 genes were down-regulated by < 0.67-fold in 93–11 (gs3/qGL3) (Fig. 1c). Comparing the transcriptomes of NIL-GS3/qgl3 (GS3/qgl3) with those of NIL-qgl3 (gs3/qgl3) and NIL-GS3 (GS3/qGL3) as well as 93–11 (gs3/qGL3), we found that 11 genes were up-regulated (Additional file 1: Table S1) and 15 genes were down-regulated (Additional file 1: Table S2). Among the 11 commonly up-regulated genes, one gene (Os03g27530) showed 18.7-fold induction under the NIL-GS3 (GS3/qGL3) background and 41.4-fold induction under the NIL-qgl3 (gs3/qgl3) background. It encoded a putative serine carboxypeptidase of the peptidase S10 family (Additional file 1: Table S1). Furthermore, we analyzed the genes commonly up- and down-regulated by qgl3 in both the NIL-qgl3 (gs3/qgl3) and NIL-GS3 (GS3/qGL3) backgrounds and found 33 up-regulated genes and 30 down-regulated genes (Additional file 1: Tables S3 and S4). By comparing the transcriptomes of the panicles of NIL-qgl3 (gs3/qgl3) and NIL-GS3 (GS3/qGL3), we found that 249 genes were up-regulated by > 1.5-fold and 237 were down-regulated by < 0.67-fold (Fig. 1c). Among these, we found a down-regulated gene, Os03g63970, encoding a GA20 oxidase involved in the GA pathway. We also discovered that some genes involved in BR signaling were differentially expressed, such as a glycogen synthase kinase (CGMC_GSK) family gene (Os05g04340) (Additional file 1: Table S6). The number of down-regulated genes was higher than the number of up-regulated genes for 93–11 and its three NILs.
To determine the identities of the differentially expressed genes (DEGs), we categorized them based on their known functions using gene ontology (GO) classifications. The DEGs between combination I (GS3/qGL3 vs. gs3/qGL3 and GS3/qgl3 vs. gs3/qgl3), combination II (GS3/qGL3 vs. GS3/qgl3 and gs3/qGL3 vs. gs3/qgl3) and combination III (GS3/qGL3 vs. gs3/qgl3) were used to analyze the GO pathways. These genes were associated with diverse biological, molecular and cellular functions, as shown in Tables 4, 5 and 6. This functional grouping primarily serves to facilitate data visualization. The functional classifications of the DEGs regulated by gs3 were mainly associated with metabolic processes, catalytic activity, and binding (Table 4). The gene Os03g27530, which is also called OsSCP16, was associated with the GO:0008152 and GO:0003824 classifications. Its homolog in Arabidopsis thaliana is BRS1, which might participate in the BR signaling pathway. Interestingly, we also found this gene in combination III. The DEGs regulated by qgl3 were mainly associated with metabolic processes, cell parts, catalytic activity, and binding (Table 5). According to q-PCR verification, the gene Os02g56310 encoding a calcium-dependent protein kinase was tremendously up-regulated in NIL-qgl3 (gs3/qgl3), NIL-GS3/qgl3 (GS3/qgl3) and 93–11 compared with NIL-GS3 (GS3/qGL3). Ca2+ sensor protein kinases are prevalent in most plant species including rice. OsCPK31, which also encodes a calcium-dependent protein kinase, played a significant role in the grain filling process and eventually reduced the crop duration in overexpression plants [33]. The DEGs regulated by both gs3 and qgl3 were associated with 51 GO terms, which included the GO terms of gs3 and qgl3 (Table 6). Of these GO terms in Table 6, many transcripts encoded proteins involved in cellular metabolic process such as NB-ARC domain containing protein, F-box domain containing protein, zinc ion binding proteins and calcium-dependent protein kinase isoform AK1. In addition to genes associated with cellular metabolic process, genes associated with Leucine-Rich-Repeat (LRR) family protein and the calcium/calmodulin depedent protein kinases were annotated with the GO term “signal transduction”. Os03g27530 and Os02g56310 were also among the DEGs regulated by gs3 and qgl3. In addition, Os07g05880 encoding F-box domain and kelch repeat containing protein, overlapping expression of rice F-box protein encoding genes during floral transition as well as panicle and seed development [34]. These results indicated that gs3 and qgl3 might participate in the same or parallel signaling pathways to regulate grain length.
Metabolic pathways, cellular response and cell regulation analysis for DEGs
To identify genes related to metabolic reconfiguration in the different combinations, the MapMan tool was used to select and display the significantly regulated metabolic pathways. From our results, the up- and down-regulated genes were classified into 36 BINs.
By MapMan analysis of the DEGs regulated by gs3, we found that most of the genes associated with the cell wall, lipids, light reactions and secondary metabolism showed down-regulation (Fig. 2a). Some genes related to the cell wall were down-regulated by gs3, implying that down-regulation of these cell wall-related genes may negatively regulate cell wall formation. In our regulation overview, protein degradation and receptor kinases were the most frequent categories (Fig. 2d). In the hormone metabolism BIN, it was found that Os03g08500 was related with ethylene synsesis. Using the cell regulation and cell response overview function of MapMan, we found that genes related to protein degradation, biotic/abiotic stress, enzyme families, and transport were highly induced (Fig. 2c). In the protein degradation BIN, four up-regulated genes (Os03g28990, Os03g39230, Os03g27530 and Os03g37950) and one down-regulated gene (Os07g05880) were involved in it. Os03g27530 was in the protein degradation BIN and might participate in the BR signaling pathway. Os03g28990 encoding a von Willebrand factor type A (vWA) domain containing protein might regulate rice vegetative growth and development. However, in the cellular response overview we only found one gene (Os03g28190) related with biotic stress (Fig. 2b). DEGs associated with the cell wall, lipids, light reactions and secondary metabolism showed up-regulation, while some genes associated with the cell wall, lipids, and ascorbate and glutathione metabolism were down-regulated by qgl3 (Fig. 3a). In the cellular response and cell regulation overview, genes related to hormones (auxin signal transduction), biotic/abiotic stress, RNA regulation of transcription, protein degradation, receptor kinase signaling, the cell cycle and protein modification were the most abundant (Fig. 3b, c). We further investigated three genes that were in the cell cycle BIN, Os02g55720, Os02g52360 and Os04g28420, all of which were up-regulated by qgl3. Os02g55720 encoded a kind of cyclin related to grain size regulation [6]. Os04g28420 encoded a kind of peptidyl-prolyl isomerase, which was up-regulated 17.97-fold by qgl3 under the NIL-gs3/qgl3 background (Additional file 1: Table S3). This indicated that qGL3 might regulate grain length through regulation of the cell cycle. The regulation overview function of MapMan showed that DEGs associated with transcription factors, protein modification, and protein degradation were significantly regulated by qgl3 (Fig. 3d). In the transcription factor BIN, it was found that some transcription factors, Os01g62130 encoding C2H2 zinc finger family protein, Os04g49450 encoding MYB related transcription and Os03g44540 encoding a CCAAT-box binding protein. The MapMan analysis indicated that some metabolic pathways were changed by allelic alterations at both loci, GS3 and qGL3 (Fig. 4a). We found that genes associated with photorespiration, light reactions, lipids, the cell wall and secondary metabolism were up-regulated, while genes related to lipids, the TCA cycle, and ascorbate and aldarate metabolisms were down-regulated (Fig. 4a). With cellular response overview, DEGs associated with biotic/abiotic stress and development were significantly regulated by both gs3 and qgl3 (Fig. 4b). DEGs in BINs such as transcription factors, protein modification, protein degradation, receptor kinases and hormones (ethylene, IAA and GA) were up-regulated by gs3 and qgl3 (Fig. 4c, d). In the GA synthesis overview, we found that a gene (Os03g63970) related with GA20 oxidase was down-regulated by both gs3 and qgl3. It is possible that BR and GA interact closely to regulate cell elongation [35]. We found that some DEGs encoded regulators, including two transcription factors, a B3 DNA binding domain-containing protein (Os03g42370) and three MYB family transcription factor (Os06g14670, Os11g47460 and Os05g51160). These regulators might take part in the same signaling pathways to increase grain length in rice, which would explain the additive effects of gs3 and qgl3 (Additional file 1: Table S5). Overall, through MapMan analysis, we found that gs3 and qgl3 were involved in some common or parallel metabolic pathways to regulate grain length.

Overview of the differentially expressed genes between GS3/qGL3 vs. gs3/qGL3 and GS3/qgl3 vs. gs3/qgl3. a Metabolism overview in MapMan. b Cellular response overview in MapMan. c Cell regulation overview in MapMan. d Regulation overview in MapMan. Red, up-regulation; white, no change; blue, down-regulation

Overview of the differentially expressed genes between GS3/qGL3 vs.GS3/qgl3 and gs3/qGL3 vs. gs3/qgl3. a Metabolism overview in MapMan. b Cellular response overview in MapMan. c Cell regulation overview in MapMan. d Regulation overview in MapMan. Red, up-regulation; white, no change; blue, down-regulation

Overview of the differentially expressed genes between GS3/qGL3 and gs3/qgl3. a Metabolism overview in MapMan. b Cellular response overview in MapMan. c Cell regulation overview in MapMan. d Regulation overview in MapMan. Red, up-regulation; white, no change; blue, down-regulation
Quantitative real-time PCR validation of DEGs
To confirm the accuracy and reproducibility of the microarray results, eight genes commonly up-regulated and six genes commonly down-regulated by gs3 and qgl3 were selected for real-time PCR verification, including five BR signaling or grain length regulation associated genes, Os11g44880, Os07g43670, Os02g56310, Os01g43890 and Os01g60280. The q-PCR results for these genes were accordance with the microarray data (Fig. 5). The eight up-regulated genes and six down-regulated genes all showed up- and down-regulation in 93–11 (gs3/qGL3), NIL-GS3/qgl3 (GS3/qgl3) and NIL-qgl3 (gs3/qgl3) compared with the NIL-GS3 (GS3/qGL3) background (Fig. 5). Strikingly, one gene, Os02g56310, encoding a calcium-dependent protein kinase, was obviously up-regulated in NIL-qgl3 (gs3/qgl3), NIL-GS3/qgl3 (GS3/qgl3) and 93–11 compared with NIL-GS3 (GS3/qGL3) (Fig. 5A).

q-PCR validation of differentially expressed genes in the four rice lines. a Eight commonly up-regulated genes. b Six commonly down-regulated genes
Discussion
Grain size is a target in breeding and natural selection, and both GS3 and qGL3 significantly regulate grain size and organ size. In this study, we compared the grain lengths of four NILs, using NIL-GS3 as a control group. The results indicated that gs3 and qgl3 had additive effects on rice grain length regulation. Moreover, qGL3 had a stronger effect on rice grain length regulation than GS3. On grain length, the strength of the additive signal from GS3 and qGL3 was much larger than the genetic interaction signal. However, there were large genetic interactions between GS3 and qGL3 on the expression levels of commonly regulated genes rather than additive effects. This work represents the first analysis of the genetic interaction between qGL3 and GS3. We used Gene Ontology [36] and MapMan [37] bioinformatics-based approaches for analyses aimed to interpret the biological significance of gene expression data. Through GO and MapMan analysis, we found that some genes regulated by gs3 and qgl3 are involved in BR signaling, the cell cycle, protein degradation, the GA/IAA family and protein modification, and might play important roles in the regulation of grain length. The gs3 up-regulated gene, Os03g27530, was in the protein degradation BIN, and its homolog (BRS1) in Arabidopsis was reported to regulate BR signaling [38]. Os05g04340 in the protein modification BIN was down-regulated by both gs3 and qgl3, and its homolog BIN2 in Arabidopsis is a negative regulator of BR signaling [39]. Based on the functional annotations of the commonly regulated genes identified in this research, the regulation of grain length by qGL3 and GS3 might involve the BR signaling pathway.
BRs are a group of steroid phytohormones ubiquitously distributed throughout the plant kingdom [23]. They have essential roles in a wide range of plant growth and development processes, and can promote cell division or elongation and enhance tolerance to environmental stresses and resistance to pathogens [40]. The signal transduction pathway of BRs has been extensively studied [39]. The phosphorylation of BSK1 (BR-signaling kinase 1) by the BR receptor kinase BR-insensitive 1 (BRI1) promotes BSK1 binding to the BRI1 suppressor 1 (BSU1) phosphatase. BSU1, in turn, inactivates the GSK3-like kinase BR-insensitive 2 (BIN2) by dephosphorylating a conserved phospho-tyrosine residue (pTyr 200) [39, 41]. qGL3 (OsPPKL1) encodes a protein phosphatase [7] and its two homologs in Arabidopsis, BSU1 and BSL1, were reported to promote brassinosteroid signaling [39, 42]. They transmit a signal by dephosphorylating and deactivating the BIN2 kinase downstream of BR signaling [39]. Moreover, we found that genes involved in BR signaling, such as the CGMC_GSK family genes, encoding Arabidopsis BIN2 homologous proteins, were differentially expressed between NIL-GS3 (GS3/qGL3) and NIL-GS3/qgl3 (gs3/qgl3). Recently, we cloned the GSK family genes and obtained additional evidence for the interaction of OsPPKL1 and GSKs via yeast two-hybrid assays (unpublished data). These data indicated that qGL3 might participate in BR signaling by dephosphorylating GSKs. However, qGL3 is a negative regulator of rice grain length [7], suggesting that OsPPKL1-GSK interaction might play different roles in BR signaling in rice compared with BSU1- and BSL1-BIN2 interaction in Arabidopsis.
GS3 is a major QTL for grain length and weight and a minor QTL for grain width and thickness [5]. GS3 was reported to be an atypical heterotrimeric G protein γ-subunit that positively regulates organ size [31, 32]. The heterotrimeric G protein α-subunit, known as D1/RGA1 in rice, is involved in an alternative BR-signaling pathway, independent of OsBRI1. Recently, Hu et al. (2013) reported that a U-Box E3 ubiquitin ligase worked as a linkage factor between the heterotrimeric Gα subunit and BR signaling to mediate rice growth, mainly by regulating cell proliferation and organizing cell files in aerial organs. In this study, we found that gs3 up-regulated a putative serine carboxypeptidase of the peptidase S10 family. Its homolog in Arabidopsis (BRS1) was reported to positively regulate BR signaling [38]. We believe that this gene might have GS5-like properties. Overexpression of BRS1 suppressed the cell surface receptor for BRs in bri1 extracellular domain mutants [38]. One of its homologs in rice was cloned as the grain-size gene GS5, which increased grain width when its expression increased [12]. These data reveal that some members of the serine carboxypeptidase family might act downstream of BR signaling as positive factors. Our research implies that GS3 also takes some part in BR signaling, and both GS3 and qGL3 might share a common BR signaling associated pathway in the regulation of rice grain length. We suppose that qGL3 might directly participate in brassinolide signaling by dephosphorylating GSKs, while GS3 indirectly influences BRS1, which is parallel to the BRI-mediated BR signaling pathway.
Among the genes up-regulated by both loci, we found a gene encoding a kinesin-4, whose homolog SRS3 was reported to positively regulate rice grain length in seed formation [25]. We identified a small and round seed mutant phenotype (srs3). The gene, which belongs to the kinesin 13 subfamily, was designated SRS3 [25]. The shortened seed phenotype of the srs3 mutant was probably the result of a reduction in cell length in the longitudinal direction [25]. The SRS3 protein might be a homolog of the AtKinesin 13A protein, which regulates trichome elongation in Arabidopsis [43]. Interestingly, among the genes commonly down-regulated by gs3 and qgl3, we observed that a number of disease resistance related genes, encoding two NB-ARC domain containing proteins, a stripe rust resistance protein Yr10 and a peroxidase precursor, were down-regulated by both qgl3 and gs3, suggesting that disease resistance responses may also be negatively correlated with grain development. In addition, we found a gene (Os07g43670) encoding a ribonuclease T2 family domain containing protein involved in the cytokinin signaling pathway. A major QTL, Grain number 1a (Gn1a), encodes a cytokinin oxidase/dehydrogenase (OsCKX2) that catalyzes the irreversible degradation of cytokinin. Mutation in Gn1a/OsCKX2 [44], which encodes a zinc finger transcription factor that directly and positively regulates Gn1a/OsCKX2 [2, 45], caused the accumulation of cytokinin and consequently increased grain number [2]. In many cases, increased grain number is closely associated with reduced grain size, likely owing to the availability of fixed carbon in the source and the efficiency of transport to the sink [7, 9, 29].
The currently available evidence suggests that the mechanisms underlying the additive effects of GS3 and qGL3 in regulating grain length might involve phytohormones (especially BRs) and key genes related to cell division or elongation. This research should help us to understand the mechanisms of the additive effects of gs3 and qgl3, which would be useful for deciphering the genetic network involved in rice seed formation and for molecular breeding.
Conclusions
With an elite indica cultivar 93–11 as recurrent parent NIL-GS3 (GS3/qGL3) and NIL-qgl3 (gs3/qgl3) were developed by conventional backcrossing and marker-assisted selection. Another line, NIL-GS3/qgl3, was developed by crossing NIL-GS3 and NIL-qgl3. By comparing the grain length of 93–11 and its three NILs we concluded that gs3 and qgl3 had additive effects on rice grain length regulation and that the effects of qGL3 were stronger. To reveal the genes affected by gs3 and qgl3, we compared the transcriptomes of the primary panicles of 93–11 and the three NILs through microarray analysis. The transcriptome analysis revealed that the genes affected by GS3 and qGL3 partially overlapped, and both loci might be involved in BR signaling.
Methods
Plant materials and development of the NILs
The high-quality, previously sequenced [46] elite indica rice cultivar 93–11 with non-functional gs3 and functional qGL3 was used as the genetic background for introducing the functional GS3 and non-functional qgl3 alleles. The japonica rice cultivar Koshihikari was used as the donor parent for functional GS3. The GS3 allele in Koshihikari was cloned and sequenced, and was found to be the same as GS3-2 (Nipponbare) [3, 18]. The rice accession N411 with extra-large grains was used as the donor parent for non-functional qgl3 [7].
As the functional GS3 is a dominant allele forming short grains, plants with 93-11-like performance with short grains were selected from BCnF1 populations of 93–11 × Koshihikari and continuously backcrossed with 93–11. To develop NIL-GS3 (genotype GS3/qGL3), we selected plants, from the BC4F1 population, with a short Koshihikari segment (from RM15144 to RM411) and the GS3 allele for self-pollination. A total of 126 simple sequence repeat markers were employed for background detection. NIL-qgl3 (genotype gs3/qgl3), which carries a ~113-kb segment, including the N411 qgl3 allele in the 93–11 background, was described in previous studies [7]. NIL-GS3/qlg3 (genotype GS3/qgl3) was developed by crossing NIL-GS3 and NIL-qgl3. In the NIL-GS3 × NIL-qgl3 F2 population, plants heterozygous at the GS3 locus and homozygous at the qgl3 locus were selected to self-pollinate naturally and the homozygous NIL-GS3/qlg3 was selected from the F3 family by MAS.
Plant growth and evaluation of agronomic traits
To evaluate the differences in grain length between the recurrent parent 93–11 and its three NILs, all materials including 93–11, NIL-qgl3, NIL-GS3 and NIL-GS3/qgl3 were grown in the Jiangpu Experiment Station of Nanjing Agricultural University. The four materials were grown in a 13.4-m2 acreage (the actual used area: 1.5 m × 8.0 m). All experimental materials were transplanted in the fields with 15 cm spacing between plants within rows and 25 cm spacing between rows. The 13.4-m2 block was divided into four plots (the area of one plot: 1.5 m × 2 m), with 80 plants of each material in one plot and there were three blocks. 10 plants selected randomly from 80 plants of each material were measured. The mean value of the 10 plants was used for analysis. T-test was carried out to evaluate the statistical differences in their grain length between NIL-GS3 and other three materials. Grain length was measured as described in a previous study [7].
Microarray analysis
As reported in previous studies, GS3 and qGL3 are expressed strongly in young panicles [4, 7]. Thus, we used primary panicles of 3–6 cm length from the three NILs and 93–11 for RNA preparation and hybridization with the Rice Genome OneArray Microarray (Phalanx Biotech Group, Hai Shang). Each NIL and 93–11 was sampled three times from different tillers. The Rice OneArray probe was set with a combination of the Rice Genome Annotation Project (RGAP) version 6.1 and Beijing Genomics Institute (BGI) version 2008 databases. Long oligonucleotide probes (~60-mers) were engineered using specific lengths to match their melting temperatures for superior hybridization performance. Each microarray contained 824 performance monitoring control probes for hybridization, sample quality, and labeling reactions. RNA isolation, purification and microarray hybridization were conducted by the Phalanx Biotech Group. Longer grains were regarded as being more active during the growth of the grain or glume. We conducted a comparison of the transcriptomes by comparing the longer grain genotype with the shorter grain genotype. The microarray data were normalized using the GC-RMA algorithm followed by Log 2 transformation. We used ordinary Student’s t –test (P value < 0.05) to identify significantly differentially expressed genes. Probe sets showing more than 1.5-fold change (four NILs) in expression were considered as DEGs. To identify DEGs regulated by gs3 or qgl3, we used the ratio (1.5 folds for up-regulation and 0.67 folds for down-regulation) of the expression level between combinations gs3/qGL3 vs. GS3/qGL3, gs3/qgl3 vs. GS3/qgl3, GS3/qgl3 vs. GS3/qGL3, and gs3/qgl3 vs. gs3/qGL3. To identify commonly expressed genes in the four materials, we used the ratio (1.5 folds for up-regulation and 0.67 folds for down-regulation) of the expression level between combination gs3/qGL3 vs. GS3/qGL3, GS3/qgl3 vs. GS3/qGL3, and gs3/qgl3 vs. GS3/qGL3. A two-way analysis of variance (ANOVA) with expression levels and genotype (GS3/gs3 and qGL3/qgl3) as main factors was applied to the datasets from all four microarrays to identify genes significantly affected by GS3, qGL3, or GS3 × qGL3 interaction. The Benjamini–Hochberg false discovery rate (FDR) for multiple test correction was used for the analysis [47]. Furthermore, the statistical criterion of at least a 1.5-fold change at a P-value ≤ 0.05 was used for gene selection.
Pathway analysis
Functional enrichment analysis of DEGs using the GO domains “molecular function”, “biological process” and “cellular component” was performed using the AGRIGO website with a significance level of FDR < 0.05 [36]. The MapMan tool [37] was employed to analyze the metabolic and signaling changes in the microarray data based on the expression value of each DEG. A metabolic pathway overview was produced by loading the DEGs with their Log2 expression values into the locally-installed MapMan program and shown using color intensity.
Real-time quantitative PCR
Based on the transcriptome comparison between the three NILs and 93–11, several DEGs were selected for further confirmation by real-time quantitative PCR. Primary panicles of 3–6 cm length were used for total RNA extraction with an RNA extraction kit (RNAiso Plus, TaKaRa Bio, Inc.). Reverse transcription was performed using 6 μg RNA and 4 μg reverse transcriptase mix (PrimeScript® RT Master Mix Perfect Real Time, TaKaRa Bio) in a volume of 40 μl, according to the manufacturer’s protocol. Real-time PCR was carried out in a total volume of 25 μl containing 2 μl of cDNA, 0.2 mM gene-specific primers, 12.5 μl SYBR® Premix Ex Taq TM II, and 0.5 μl of Rox Reference Dye II (TaKaRa Bio), using an ABI 7500 Fast Real-Time PCR System according to the manufacturer’s instructions. The rice 18S rRNA gene was used as an internal control. Relative quantification of the transcript levels was performed using the 2−ΔΔCT method [48].
Availability of supporting data
The microarray data for the four NILs has been submitted to the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE59619.
Abbreviations
- NIL:
-
Near-isogenic lines
- MAS:
-
Marker-assisted selection
- QTL:
-
Quantitative trait locus
- SSR:
-
Simple sequence repeat
- ANOVA:
-
Analysis of variance
- GL:
-
Grain length
- DEG:
-
Differentially expressed gene
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- GO:
-
Gene ontology
- FDR:
-
False discovery rate
References
Xing YZ, Zhang QF. Genetic and Molecular Bases of Rice Yield. Annu Rev Plant Biol. 2010;61:421–42.
Zuo J, Li J. Molecular Genetic Dissection of Quantitative Trait Loci Regulating Rice Grain Size. Annu Rev Genet. 2014. DOI: 10.1146/annurev-genet-120213-092138.
Mao HL, Sun SY, Yao JL, Wang CR, Yu SB, Xu CG, et al. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci U S A. 2010;107(45):19579–84.
Takano-Kai N, Jiang H, Kubo T, Sweeney M, Matsumoto T, Kanamori H, et al. Evolutionary History of GS3, a Gene Conferring Grain Length in Rice. Genetics. 2009;182(4):1323–34.
Fan CH, Xing YZ, Mao HL, Lu TT, Han B, Xu CG, et al. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet. 2006;112(6):1164–71.
Qi P, Lin YS, Song XJ, Shen JB, Huang W, Shan JX, et al. The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1;3. Cell Res. 2012;22(12):1666–80.
Zhang X, Wang J, Huang J, Lan H, Wang C, Yin C, et al. Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc Natl Acad Sci U S A. 2012;109(52):21534–9.
Ishimaru K, Hirotsu N, Madoka Y, Murakami N, Hara N, Onodera H, et al. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat Genet. 2013;45(6):707–11.
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–30.
Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, et al. Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet. 2008;40(8):1023–8.
Weng JF, Gu SH, Wan XY, Gao H, Guo T, Su N, et al. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 2008;18(12):1199–209.
Li Y, Fan C, Xing Y, Jiang Y, Luo L, Sun L, et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet. 2011;43(12):1266–9.
Wang SK, Wu K, Yuan QB, Liu XY, Liu ZB, Lin XY, et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet. 2012;44(8):950–4.
Xie XB, Song MH, Jin FX, Ahn SN, Suh JP, Hwang HG, et al. Fine mapping of a grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon. Theor Appl Genet. 2006;113(5):885–94.
Guo LB, Ma LL, Jiang H, Zeng DL, Hu J, Wu LW, et al. Genetic Analysis and Fine Mapping of Two Genes for Grain Shape and Weight in Rice. J Integr Plant Biol. 2009;51(1):45–51.
Bai XF, Luo LJ, Yan WH, Kovi MR, Zhan W, Xing YZ: Genetic dissection of rice grain shape using a recombinant inbred line population derived from two contrasting parents and fine mapping a pleiotropic quantitative trait locus qGL7. Bmc Genet 2010;11(16):2187.
Shao GN, Tang SQ, Luo J, Jiao GA, Wei XJ, Tang A, et al. Mapping of qGL7-2, a grain length QTL on chromosome 7 of rice. J Genet Genomics. 2010;37(8):523–31.
Shao GN, Wei XJ, Chen ML, Tang SQ, Luo J, Jiao GA, et al. Allelic variation for a candidate gene for GS7, responsible for grain shape in rice. Theor Appl Genet. 2012;125(6):1303–12.
Qiu X, Gong R, Tan Y, Yu S. Mapping and characterization of the major quantitative trait locus qSS7 associated with increased length and decreased width of rice seeds. Theor Appl Genet. 2012;125(8):1717–26.
Ashikari M, Wu JZ, Yano M, Sasaki T, Yoshimura A. Rice gibberellin-insensitive dwarf mutant gene Dwarf 1 encodes the alpha-subunit of GTP-binding protein. Proc Natl Acad Sci U S A. 1999;96(18):10284–9.
Izawa Y, Takayanagi Y, Inaba N, Abe Y, Minami M, Fujisawa Y, et al. Function and Expression Pattern of the alpha Subunit of the Heterotrimeric G Protein in Rice. Plant Cell Physiol. 2010;51(2):271–81.
Oki K, Inaba N, Kitano H, Takahashi S, Fujisawa Y, Kato H, et al. Study of novel d1 alleles, defective mutants of the alpha subunit of heterotrimeric G-protein in rice. Genes Genet Syst. 2009;84(1):35–42.
Tanaka A, Nakagawa H, Tomita C, Shimatani Z, Ohtake M, Nomura T, et al. BRASSINOSTEROID UPREGULATED1, Encoding a Helix-Loop-Helix Protein, Is a Novel Gene Involved in Brassinosteroid Signaling and Controls Bending of the Lamina Joint in Rice. Plant Physiol. 2009;151(2):669–80.
Abe Y, Mieda K, Ando T, Kono I, Yano M, Kitano H, et al. The SMALL AND ROUND SEED1 (SRS1/DEP2) gene is involved in the regulation of seed size in rice. Genes Genet Syst. 2010;85(5):327–39.
Kitagawa K, Kurinami S, Oki K, Abe Y, Ando T, Kono I, et al. A Novel Kinesin 13 Protein Regulating Rice Seed Length. Plant Cell Physiol. 2010;51(8):1315–29.
Segami S, Kono I, Ando T, Yano M, Kitano H, Miura K, Iwasaki Y: Small and round seed 5 gene encodes alpha-tubulin regulating seed cell elongation in rice. Rice 2012;5(4):225.
Nakagawa H, Tanaka A, Tanabata T, Ohtake M, Fujioka S, Nakamura H, et al. Short grain1 decreases organ elongation and brassinosteroid response in rice. Plant Physiol. 2012;158(3):1208–19.
Ying JZ, Gao JP, Shan JX, Zhu MZ, Shi M, Lin HX. Dissecting the genetic basis of extremely large grain shape in rice cultivar ‘JZ1560’. J Genet Genomics. 2012;39(7):325–33.
Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, et al. Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet. 2009;41(4):494–7.
Zhou Y, Zhu J, Li Z, Yi C, Liu J, Zhang H, et al. Deletion in a quantitative trait gene qPE9-1 associated with panicle erectness improves plant architecture during rice domestication. Genetics. 2009;183(1):315–24.
Li S, Liu W, Zhang X, Liu Y, Li N, Li Y. Roles of the Arabidopsis G protein gamma subunit AGG3 and its rice homologs GS3 and DEP1 in seed and organ size control. Plant Signal Behav. 2012;7(10):1357–9.
Li S, Liu Y, Zheng L, Chen L, Li N, Corke F, et al. The plant-specific G protein gamma subunit AGG3 influences organ size and shape in Arabidopsis thaliana. New Phytol. 2012;194(3):690–703.
Manimaran P, Mangrauthia SK, Sundaram RM, Balachandran SM. Constitutive expression and silencing of a novel seed specific calcium dependent protein kinase gene in rice reveals its role in grain filling. J Plant Physiology. 2015;174:41–8.
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, et al. F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 2007;143(4):1467–83.
Tong H, Xiao Y, Liu D, Gao S, Liu L, Yin Y, et al. Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. The Plant cell. 2014;26(11):4376–93.
Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic acids research. 2010;38(Web Server issue):64–70.
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37(6):914–39.
Li J, Lease KA, Tax FE, Walker JC. BRS1, a serine carboxypeptidase, regulates BRI1 signaling in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2001;98(10):5916–21.
Kim TW, Guan SH, Sun Y, Deng ZP, Tang WQ, Shang JX, et al. Brassinosteroid signal transduction from cell-surface receptor kinases to nuclear transcription factors. Nat Cell Biol. 2009;11(10):1254–60.
Zhang C, Bai MY, Chong K. Brassinosteroid-mediated regulation of agronomic traits in rice. Plant Cell Rep. 2014;33(5):683–96.
Kim TW, Wang ZY. Brassinosteroid Signal Transduction from Receptor Kinases to Transcription Factors. Annual RevPlant Biol. 2010;61:681–704.
Mora-Garcia S, Vert G, Yin Y, Cano-Delgado A, Cheong H, Chory J. Nuclear protein phosphatases with Kelch-repeat domains modulate the response to brassinosteroids in Arabidopsis. Gene develop. 2004;18(4):448–60.
Lu L, Lee YRJ, Pan RQ, Maloof JN, Liu B. An internal motor kinesin is associated with the golgi apparatus and plays a role in trichome morphogenesis in Arabidopsis. Mol Biol Cell. 2005;16(2):811–23.
Ashikari M, Sakakibara H, Lin SY, Yamamoto T, Takashi T, Nishimura A, et al. Cytokinin oxidase regulates rice grain production. Science. 2005;309(5735):741–5.
Li SY, Zhao BR, Yuan DY, Duan MJ, Qian Q, Tang L, et al. Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc Natl Acad Sci U S A. 2013;110(8):3167–72.
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science. 2002;296(5565):79–92.
Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57(1):289–300.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods. 2001;25(4):402–8.
Acknowledgements
The authors thank Mr. Congfei Yin and Dr. Yunyu Wu for their experimental help. This work was supported by grants from the Natural Science Foundation of China (Grant Nos. 31071386 and 91335106) and the Fundamental Research Funds for the Central Universities (No. KYZ201137).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HSZ, XJZ and JFW designed the research. XYG, XJZ and HXL constructed the four NILs. XJZ and XYG analyzed the microarray data. JH analyzed the metabolic and signaling changes with the microarray data. XYG performed quantitative real-time PCR. All authors read and approved the final manuscript.
Xiuying Gao and Xiaojun Zhang contributed equally to this work.
Additional file
Additional file 1: Table S1.
Genes up-regulated by gs3 (>1.5-fold). Table S2. Genes down-regulated by gs3 (<0.67-fold). Table S3. Genes up-regulated by qgl3 (>1.5-fold). Table S4. Genes down-regulated by qgl3 (<0.67-fold). Table S5. Genes up-regulated by both qgl3 and gs3 (<0.67-fold). Table S6. Genes down-regulated by both qgl3 and gs3 (<0.67-fold). Additional file 1: Table S7. qGL3 × GS3 interactions resolved by two-way ANOVA for the expression level of commonly regulated genes.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Gao, X., Zhang, X., Lan, H. et al. The additive effects of GS3 and qGL3 on rice grain length regulation revealed by genetic and transcriptome comparisons. BMC Plant Biol 15, 156 (2015). https://doi.org/10.1186/s12870-015-0515-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-015-0515-4