
Abstract
Background
Members of Paenibacillus genus from diverse habitats have attracted great attention due to their multifarious properties. Considering that members of this genus are mostly free-living in soil, we characterized the genome of a halotolerant environmental isolate belonging to the genus Paenibacillus. The genome mining unravelled the presence of CAZymes, probiotic, and stress-protected genes that suggested strain S-12 for industrial and agricultural purposes.
Results
Molecular identification by 16 S rRNA gene sequencing showed its closest match to other Paenibacillus species. The complete genome size of S-12 was 5.69 Mb, with a GC-content 46.5%. The genome analysis of S-12 unravelled the presence of an open reading frame (ORF) encoding the functions related to environmental stress tolerance, adhesion processes, multidrug efflux systems, and heavy metal resistance. Genome annotation identified the various genes for chemotaxis, flagellar motility, and biofilm production, illustrating its strong colonization ability.
Conclusion
The current findings provides the in-depth investigation of a probiotic Paenibacillus bacterium that possessed various genome features that enable the bacterium to survive under diverse conditions. The strain shows the strong ability for probiotic application purposes.
Similar content being viewed by others


Background
Paenibacillus genus was observed in 1993 with an estimated 11 species belonging to the genus Bacillus [1]. Many new members of this genus have been discovered and so far, more than 150 species have been identified. The identified members are endospore-forming, facultative anaerobic, rod-shaped, and motile [2, 3]. The spore forming ability helps Paenibacillus species to persist for a long period, which makes it a unique bacterium as compared to other bacterial strains in the environment [4, 5]. The reported members are morphologically, physiologically, and biochemically diverse and are present in environments like water [6], soil [7], insect larvae [8], and human feces [3]. Initially, members of this genus were reported to enhance plant growth such as Paenibacillus polymyxa [9]. Recently Paenibacillus strains showed their ability for silver nanoparticle production [10], ginsenoside transformation [11], and ginsenoside Rd production [12].
Besides their ability to plant protection, lipopetide producing Paennibacillus spp. has achieved great attention for the treatment of drug-resistant and human bacterial pathogens [13]. Similarly, tridecaptins produced by Paenibacillus spp. showed strong inhibitory activity against multidrug-resistant bacterial strains by binding to lipid II on the bacterial inner membrane leading to disruption of proton motive force [14]. The produced tridecaptins showed low cytotoxicity and higher stability in human plasma. The other member P. larvawas identified as spreading epizootic disease by American Foulbrood (AFB) [8]. These bacteria were equipped with various carbohydrate (cellulose, starch, and xylan) hydrolyzing enzymes [15, 16]. Genome analysis identified the genes for antimicrobial and volatile organic compounds, siderophore production, phosphate transport, and indole acetic acid (IAA) synthesis etc. in P.yonginensis DCY84T [17].
Among the reported Paenibacillus strains, P. polymyxa showed probiotic features. The livestock feed isolated bacterium P. polymyxa JB-0501 was exploited as a potent probiotic candidate following in-vitro evaluation [18]. Another bacterium P. polymyxa strain HGA4C isolated from fish gut demonstrated the antibacterial and probiotic features under in-vitro and in-vivo conditions [19]. The bacterium Paenibacillus sp. Aloe-11 secretes the extracellular enzymes for metabolization of complex polysaccharides and colonized the chicken intestine [20]. A strain of P. polymyxa is marketed as a potent probiotic for aquaculture purposes under the Biostart® trade name [21].
Besides genome sequencing, comparative genomics provides valuable information on the gene repertoires associated with metabolic activities and their adaptability to the environment [22, 23]. Additionally, comparative genomics tells us about other processes like gene duplication, increased flux expansion through horizontal gene transfer (HGT), gene loss, and genome reduction [24]. Members of the Paenibacillus genus showed potent environmental adaptability and a broad genome size [25, 26]. However, the detailed information about the adaptability of this genus based on the genome dynamics is still unclear. Till now, more than 100 Paenibacillus genomes have been sequenced, which require in-depth analysis to understand the gene repertories linked with adaptability to diverse environments.
Several features like bacterial colonization [27], antagonistic activity [28] and the ability to confer induced resistance [29] are the main reported mechanism of biological control. Previous studies [30, 31] showed that P. polymyxa secretes various metabolites including polymyxins, fusaricidins, and other antibiotics belonging to the category of antibacterial and antifungal activities. The different types and amounts of antibiotics affect the antimicrobial spectra and their biocontrol efficacy. The biocontrol ability might be due to the release of various volatile organic compounds mainly 2,3-butanediol, isoprene, butyl acetate, and n-hexadecane etc. into the surrounding environment which imparts a protective effect on plants [29, 32]. Some of the Paenibacillus spp. is well known for their ability to induce plant growth [8, 9]. Recently, Furlan et al. reported a polyethylene degrading bacterium P. aquistagni strain DK1 and identified an alkB-like gene and other structural motifs related to alkane hydrolases such as His boxes, and HYG motif [33].
In the last few decades, biosynthetic gene clusters (BGCs) related to secondary metabolite production has been characterized in microbes [34]. Identification of these BGCs explores the information about the genes encoding key signature enzymes and antimicrobials proteins (AMPs) [34]. These AMPs include polymyxin, paenibacterin, and lipopeptides which showed strong inhibitory activity against bacteria, fungi and even cancer cells [35, 36]. These BGCs includes the NRPSs (nonribosomal peptide synthetases), PKSs (polyketide synthase) and RiPPs (ribosomally synthesized and post-translationally modified peptides) [37]. The exploration of BGCs in microbes provides important information about the distribution of various genes for secondary metabolites production and the identification of potent industrially relevant strains with novel and/or improved functionality [38]. This information can be used for developing industrial-relevant strains for their wide applications. The secondary metabolites are required for the growth and development of the host as well as confer protection against infections [39]. Considering the great demand for the identification of new bacterial strains with the ability to control the growth of pathogenic microbes, we explored the genome annotation of Paenibacillus sp. S-12 to identify BGCs and AMPs. The use of AMPs in the food industry as natural antimicrobial agents is generally recognized as safe (GRAS) and promises safety and food product quality [40, 41].
Carbohydrate-active enzymes (CAZymes) constitute broader enzyme group involved in the degradation as well as rearrangement of glycosidic bonds in carbohydrates [42]. These CAZymes are classified majorly into glycosyl transferases (GTs), glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), and auxillary activities (AAs) [43]. Previously, CAZymes were noted by the genome annotation of Paenibacillus sp. JDR-2, P. mucilaginous KNP414, and P. terrae HPL-003. Recently, carbohydrate-active enzymes (CAZymes) as well as enzymes responsible for woody biomass degradation in termite gut were identified in the genome of P. polymyxa A18 [44]. However, the increasingly industrial applications of CAZymes demand the exploration of new microbial resources for more diverse CAZymes.
The gene repertoires of the genus Paenibacillus are in incessant flux and the genome size of Paenibacillus shows high plasticity, therefore, we explored the genomic analysis of newly isolated bacterium Paenibacillus sp. S-12. The aim of the work was to carry out the physiological, biochemical and genomic characterization of halotolerant Paenibacillus sp. S-12. The strain possesses several beneficial gene features such as presence of CAZymes, which make it an important microorganism for use in the area of environmental biotechnology and other industrial applications.
Methodology
Bacteria isolation
The bacterial strain S-12 was isolated from the rhizospheric soil of Rauvolfia serpentine growing around the salt belt region of Ranchi, India (23.41° N, 85.43° E). The attached soil was brought to the laboratory in ziplog bags and serially diluted in 1X PBS (phosphate buffer saline) solution. The serially diluted samples were plated on the sterile LB-agar plate amended with different salt concentrations (2 to 8% NaCl) and incubated at 37 °C overnight. One colony showing luxuriant growth on 6% salt-amended plate was further used for detailed characterization. Glycerol stock (20% v/v) was used for the preservation of bacterial culture.
Biochemical characterization
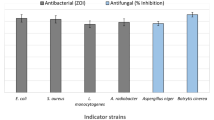
Gram staining of strain S-12 was done by bacterial Gram-stain kit (Himedia, India). The strain S-12 was tested for biochemical characteristics such as IMViC (Indole, Methyl Red, and Voges Proskauer), citrate utilization, catalase and oxidase activity following the standard protocol. The strain was tested for pH tolerance (pH 5 to 10) and temperature tolerance (25 ℃ to 45 ℃). The capacity of utilizing different carbon sources was tested by KB009-HiCarbo Kit (Himedia, India). For the carbon utilization test, strain S-12 was grown in tryptic soya broth (Himedia, India) up to 0.6 at 600 nm. Following growth, 50 µl of bacterial suspension was aseptically transferred into each well and kept for incubation for 24 to 48 h at 37 ℃. The interpretation of results was recorded following observation of colour change and as per the instruction sheet supplied with the kit. The antagonistic activity against bacterial strains (E. coli, P. aeruginosa, B. subtilis, S. aureus), and fungal strains (Aspergillus niger, Microsporum gypseum, H. gypsium and Penicillium citrium) was performed by well diffusion method [45].
Antibiotics sensitivity test
The strain S-12 was tested for their susceptibility against diverse antibiotics namely erythromycin (15 µg), ampicillin (10 µg), kanamycin (30 µg), tetracycline (30 µg), ciprofloxacin (5 µg), gentamicin (10 µg), fluconazole (25 µg), streptomycin (10 µg), vancomycin (30 µg), and voriconazole (15 µg) following CLSI (Clinical and Laboratory Standards Institute) instruction. The plates were incubated for 24–48 h at 37 °C and the result was interpreted by measuring the zone of inhibition (ZOI).
Scanning electron microscopy
Scanning electron microscopy (SEM) was performed to observe the morphology of the test isolate. The overnight grown culture was pellet down in 2 ml Eppendorf tubes and washed three times with PBS (phosphate buffer solution). The pellet was fixed in the mixture of glutaraldehyde and buffer in the ratio of 1:9 at pH 7.2. After mixing, the sample was kept on ice for 45–60 min. The suspension was centrifuged and the pellet was dehydrated in ascending grades of ethyl alcohol. The dried sample was transferred in a carbon stub and further moved for SEM analysis (JSM-6390LV, Jeol, Japan) at 500-1000X magnification.
Molecular identification
The genomic DNA of S-12 was extracted from the mid-exponential phase using Qiagen Kit (Qiagen, Germany). The S-12 strain was identified as Paenibacillus sp. by amplification and sequencing of the 16 S rRNA gene using the universal 27 F1 and 1492 R2 primers set [46] .The condition for PCR includes an initial denaturation at 95 °C for 5 min, followed by 30 cycles at 95 °C for 30 s, 55 °C for 40 s, and 72 °C for 40 s, with the final step at 72 °C for 10 min. The PCR amplification was performed in a Gene-Amp PCR system 2400 (Applied Biosystem, Thermo Scientific, USA). The amplified product was subjected to 1% gel electrophoresis, stained with ethidium bromide and the product was sequenced at Eurofins Genomic Labs Ltd. (Eurofins, India). The obtained sequence was analyzed for BLAST homology at the public available NCBI database http://www.ncbi.nlm.nih.gov/BLAST. The Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo) was sued for sequence alignment and the phylogenetic tree was constructed by MEGA 7.0 [47]. The 16 S rRNA sequence was submitted to the Genbank database for obtaining an accession number.
Screening for probiotic propensities
The test isolate S-12 was grown in the TSB (tryptic soya broth) (Himedia, India) and late log phase cells were collected by centrifugation at 8,000 rpm for 15 min at 4 ℃. The obtained cells were washed with sterile 1X PBS buffer (pH 7.4) and adjusted to a cell density of 1 × 107CFU/ml. The acid tolerance was tested by inculcating in the sterile TSB medium with different pH (2.0 & 7.0) and kept for incubation at 37 ℃ for 90 min. The cells count was performed by plating the 100 µl of serially diluted cell suspension on sterile TSA (tryptic soya agar) plates. Similarly, bacterial cells were inoculated into the TSB medium supplemented with 0.2%, 0.3% and 0.4% bile (Himedia, India) to test the bile tolerance [48]. Following, incubation at 37 ℃ for 24 h, the viability was assessed as mentioned above. The medium without bile was used as a control.
Biofilm formation test
The biofilm formation ability of strain S-12 was tested by the standard crystal violet (CV) staining method [49]. The bacterial population of S-12 was adjusted to 108CFU/ml in the TSB medium and cell suspension of 20 µl was transferred to a 96-well microtiter plate filled with 180 µl of sterile LB (Himedia, India). The control wells were filled with LB-medium only. The plates were incubated at 37 °C for 24 h under static conditions. To check the biofilm formation ability, separate plates with the above-mentioned treatment were incubated at 25, 30, 35, and 40 °C. Following incubation, medium was discarded and wells were washed with 3x PBS and filled with 200 µl of 0.1% CV (Sigma-Aldrich, USA), and incubated at room temperature for 30 min. The CV was decanted and fixing of bacteria was done by hot air stream at 60 ℃ for 1 h. The resolubilization of dye-bound cells was performed by three rinses with 200 µl of ethanol (Merck, India) per well and OD was measured at 595 nm using a microtiter plate reader. Each treatment was done in triplicate to calculate the average value and for accuracy the experiment was repeated three times.
Endospore formation test
Endospore formation ability of S-12 was evaluated microscopically and by standard plating procedure. The test strain was grown in Difco sporulation medium (DSM: Difco nutrient broth 8 g/l, MgCl2 0.49 mM, KCl 13.4 mM, Ca(NO3)2 1 mM, MnCl2 10 µM, and FeSO4 1µM) for 48 h at 37 °C with constant stirring at 180 rpm. Bacteria was pelleted at 10,000 rpm for 10 min at 4 °C and washed in cold-sterile water followed by heat shock at 70 °C for 30 min to kill the vegetative cells. The sporulating cells were observed under phase-contrast microscopy.
Genome sequencing, assembly and annotation
Genomic DNA was isolated from the QIAamp DNA Mini Kit (Qiagen, Germany). The extracted DNA sample was sequenced using an illumine MiSeq platform and the paired-end library was prepared by using the NEB Next Ultra DNA Library Prep Kit. Fast QC program was used for Quality control of Illumina reads (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The Illumina reads were used for hybrid assembly using the SPAdes version 3.15.2 [50].The transfer RNA (tRNA) and ribosomal RNA (rRNA) of the S-12 were identified using the tRNAscan-SE and RNAmmer (v1.2, http://www.cbs.dtu.dk/services/RNAmmer/) software, respectively. The genome was annotated using Prokka [51] and RAST [52] tool against the NCBI non-redundant (NR) database. The COG (clusters of orthologous groups) analysis of protein was performed by BLASTP [53]. The genes involved in biological pathways were annotated using KEGG and Blast2Go tools [54]. The analysis of orthologous gene clusters was analyzed using the Orthovenn2 program [55] using protein sequences of S-12, P. alvei DSM29, P. curdlanolyticus YK9, P. polymyxa ATCC842, P. polymyxa E681, and P. polymyxa SC2. The multiple alignments of the genome sequences were performed using MUSCLE [56]. The tree was constructed by NJ-method following the kimura-2 model using the MEGA 7.0 [57, 58].
Antimicrobial and virulence analysis
The CARD database was used using a homology based approach (BLASTX) against the genome sequence of S-12 to unravel the presence of AMR genes. For searching, BLAST output was filtered with a minimum 70% identity and subject protein coverage. Similarly, the VFDB database was used against assembled genome with criteria of a minimum of 70% identity using a homology-based approach (BLASTX) to identify the virulence genes.
Detection of secondary metabolite biosynthetic gene cluster
The number and types of secondary metabolite BGCs in the genome sequence of S-12 was identified by antiSMASH version 5.1.2 in combination with Hidden Markov Model (HMM) to detect the BGCs-like region [37, 59]. Various unknown and characterized BGCs were identified and genetic similarities in gene clusters were predicted using antiSMASH 5.1.2.
Prediction of carbohydrate‑active enzyme (CAZymes)
To reveal the presence of various CAZymes including glycosyltransferases (GTs), glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrateesterases (CEs), auxiliary activities (AAs), and carbohydrate-binding modules (CBMs), the protein sequences of S-12 was annotated using the dbCAN2 server [60] and BLAST-driven DIAMOND against the CAZy database [43]. The diversity of CAZymes in closest relatives of Paenibacillus species was also performed to evaluate the comparative distribution.
Pan genome-core genome analysis
Strain S-12 and its closest non-type strains were used for prediction of core and accessory genes using Roary v 3.11.2 with default setting [61] based on GFF3 file of all selected genome generated through PROKKA v 1.14.5 [51]. The strains were selected based on the higher similarity of S-12 to other closely related strains in the RAST (Rapid Annotation using Subsystem Technology). To determine the presence and absence of each core gene in selected strains, matrix was visualized with Phandango [62].
Results
Biochemical characterization
The isolated strain was found to be a Gram-positive, rod shaped bacterium. Among biochemical tests, it showed the negative result for indole, methyl-red and positive for voges-proskauer, citrate, oxidase and catalase (Table S1). The strain was able to grow in a wide range of pH (5 to 9) and temperature (30 to 40 ℃) (Table S1). Among the tested various carbon sources, the strain was able to utilize lactose, xylose, maltose, galactose, melibiose, sorbitol, glycerol, D-arabinose, citrate, malonate, ONPG, inulin, inositol, trehalose and mannoside etc. (Supplementary Table 1). The isolate showed a higher sensitivity (20 to 25 mm) against streptomycin, gentamicin, erythromycin, ciprofloxacin, vancomycin, and moderate sensitivity (10 to 18 mm) to ampicillin, fluconazole, tetracycline, and resistant to kanamycin, voriconazole (Supplementary Table 2). SEM analysis confirms the rod-shaped colony of the isolated bacterium (Supplementary Figure S1). Phenotypically on a motility-specific medium, the strain showed the swimming, swarming, and twitching motility behavior (Supplementary Figure S2). The test isolate showed good antagonistic activity against E. coli, P. aeruginosa, and moderate against B. subtilis and S. aureus. Against the fungal strains, strain showed good antagonistic activity against A. niger, M. gypseum, and moderate against H. Gypsium and P. citrium (Supplementary Table 3).
Identification and phylogenetic analysis
For identification at molecular level, 1.5 Kb PCR amplicon of 16 S rRNA gene was sequenced at Eurofins Genomics Pvt Ltd. (Karnataka, India), and obtained sequence was submitted to the NCBI Genbank (accession no. OM943075). Following BLAST result, the strain was identified as Paenibacillus sp. with the closest match of 98% similarity to Paenibacillus sp. 4RB2 (Fig. 1). The strain also showed its closest similarity to other Paenibacillus strains used in this study.
Probiotic properties
The selection of probiotic strain involves its ability to sustain the low pH of the stomach as well as the presence of bile in the intestine. The test isolate exhibited good tolerance to low pH and bile salts. In response to low pH, there was a decrease in bacterial counts from 1.0 × 107 to 4.5 × 104 (Supplementary Figure S3). Following exposure to bile salts, there was no significant decrease in the viability of S-12 (Supplementary Figure S3). The genome analysis revealed the presence of genes responsible for pH homeostasis and metabolic rearrangements, general and secondary stress, which ensures its survival during gastrointestinal transit. In the S-12 genome, we identified the genes for Na+/H+ antiporter, F0/F1-ATP synthase, alcohol and lactate dehydrogenase, and amino acid decarboxylase which assist the bacteria in stress survival. Additionally, arcD genes corresponding to arginine/ornithine antiporters, glutamate decarboxylase, and arginine deaminase were also identified (Table 1). The various other proteins involved in the repair of macromolecules such as DnaK/DnaJ chaperone, GroEL, and GroES were identified in the S-12 genome.
Biofilm formation ability
The test strain S-12 was identified as biofilm producers. The isolate displayed a moderate biofilm formation ability (OD595 < 0.462) at 25 °C, whereas at 30, 35, and 40 °C, it showed good biofilm (OD595 > 0.462) formation (Supplementary Figure S4). Paenibacillus S-12 formed mature spores in the DSM medium and microscopic observation showed the DPA accumulation in the centre (Supplementary Figure S5).
Genome analysis
The genome sequencing of S-12 using the Illumina sequencing platform generated 1,230,695,800 bp paired-end reads. The de-novo assembly of the reads using SPAdes generated twenty seven contigs constituting a circular chromosome of 5.69 Mb (Fig. 2). The genome annotation noted 6068 protein coding genes (CDSs), 27 rRNA, and 15 tRNA in the genome of S-12 (Table 2).Orthovenn2 exhibited that protein clusters shared by all six species were 2082, 308 shared by five species, 1651, 303 and 2826 shared by four, three, and two species, respectively. A total of 465 protein clusters were specific to only a single genome (Fig. 3a). Out of the 465 gene clusters, 274 belonged to Paenibacillus sp. S-12, 139 to P. alvei DSM29, 25 to P. polymyxa ATCC 842, and 27 to P. polymyxa SC2. Protein coding gene comparison was performed between Paenibacillus sp. S-12 and other five closely related strains. The first pattern shows the gene clusters in the graph, whereas clusters count and total protein count are displayed in second and third pattern of the stacker graph, respectively (Fig. 3b). Similarly, the heat map between S-12 and the other five strains demonstrated overlapping gene clusters in a pair wise pattern (Fig. 3c). The lowest thresholds of gene clusters were observed between Paenibacillus sp. S-12 and P. alvei DSM 29.
RAST functional annotation
Genes were predicted in the S-12 genome using RAST (http://rast.nmpdr.org/) server and subsystem/non-subsystem coverage generated are 46% and 54%, respectively (Fig. 4). The top three subsystem category distributions are amino acids and derivatives (1021 genes), carbohydrate (964 genes) and protein metabolism (564 genes). The other subsystem categories of vitamins & cofactors (546 genes), RNA metabolism (389 genes), cell wall and capsules (364 genes), and fatty acid metabolism (345 genes) were observed (Fig. 4).
RAST-based functional annotation identified the various genes associated with flagellar motor protein, flagellar biosynthesis, and chemotaxis. Among flagellar biosynthesis, we observed the genes FlhA/B and Flil for the flagellar structure formation. Among flagellar motor proteins, genes responsible for flagellar motor rotation protein MotA/B, and genes for flagellar motor switch protein FliM/N were observed. Similarly, chemotaxis-associated genes such as CheA/V/Y were identified (Supplementary Table 4). Among multidrug resistance efflux pumps, RND family MDR membrane protein CmeA/B, outer membrane lipoprotein CmeC, transcription repressor of multidrug efflux pump belonging to acrAB operon, and TetR (AcrR) family were observed. Similarly, the MATE family of MDR efflux pumps belonging to extrusion protein (Na+/drug antiporter), toxin extrusion pump YdhE/NorM, and Multidrug-efflux superfamily (MFS) transporter was also observed (Supplementary Table 5).
Genome annotation also identified the various stress-tolerant genes like glutathione redox reaction (15), CoA-disulfide thiol-di-sulfide redox system (1), redox-dependent regulatory proteins (13), rubrerythrin (40), cold shock CspA family protein (8), and DNAK family (33) genes. The other genes include the detoxification stress response (31), flavohaemoglobin (5), sigma B stress response regulation (9), Hfl operons (4), and carbon starvation (8). Various genes related to iron uptake like siderophore enterobactin (2), bacillibactin siderophore (13), iron siderophore, sensor and receptor system (3), and siderophore anthrachelin (7) were identified.
Gene ontology
COG analysis predicted the highest number of genes (16) for the ABC-type multidrug transport system, followed by the DNA-binding response regulator (OmpR family, 14) and permease component (12) (Fig. 5a). An equal number of genes (11) belonging to DNA directed RNA polymerase and AcrR family of DNA-binding transcriptional regulators were identified. Similarly, equal numbers (10) of maltose binding protein MalE and beta-lactamase class C family proteins were also observed. It was followed by the equal number of genes (10) for MFS family efflux permease and signal transduction histidine kinase. KEGG analysis showed the various proteins belonging to different metabolic pathways (Fig. 5b). The highest number was recorded for metabolic pathways (390), biosynthesis of secondary metabolites (125) followed by genes (115) responsible for metabolism in diverse environments.
AMR and VF analysis
We observed various AMR genes belonging to the category of major facilitator superfamily (MFS) antibiotic efflux pump, small multidrug resistance (SMR) antibiotic efflux pump, fluoroquinolone-resistant parC, resistance-nodulation-cell division (RND) antibiotic efflux pump and ADC beta-lactamase. Among the different drug class, we observed the genes for macrolide antibiotic, fluoroquinolone antibiotic, cephalosporin, tetracycline antibiotic, fluoroquinolone antibiotic, aminocoumarin antibiotic, lincosamide antibiotic, streptogramin antibiotic, and fosfomycin. The different antibiotic resistance genes with their functional class have been summarized in Supplementary Figure S6 a&b. The VFDB analysis identified various genes related to virulence characteristics such as adherence, invasion, exotoxin, biofilm formation, and transporters etc. (Fig. 6). The highest percentage was noted for adherence (31%), transporters and regulation (17%), iron uptake (10%), exotoxin (9%), and biofilm (7%) etc.
Genomic island (GI)
Using Island viewer, GI was identified in the S-12 genome. Predicted GI of Paenibacillus sp. S-12 includes various hypothetical proteins, separation proteins, peptidase, and survival proteins (surA) which assist the bacteria in survival in diverse environments (Supplementary Figure S7).
Biosynthetic gene clusters
The antiSMASH analysis identified genes for antimicrobial peptides (AMPs), secondary metabolite production, NRPSs, and PK synthesis. Various NRPs regions related to paenibacterin, guadinomine, polymyxin B, chejuenolide A/ chejuenolide B, fusaricidin, pelgipeptin, and octapeptin-C4 were identified (Fig. 7). The gene cluster for arylpolyene, siderophore like staphylobactin, lanthipeptide-class of S-layer protein, and betalactone were observed (Fig. 7). Fengycin and paenibacterin are the lipopolysaccharides (LPs) identified through antiSMASH analysis.
CAZy carbohydrase analysis
The CAZy analysis revealed that S-12 has 95 genes for GHs, 62 for GTs, 37 for CEs, and 21 belonging to AAs, whereas 19 and 4 were related to CBMs and PLs, respectively (Table 3). In the GHs group, the higher subcategory was observed for GH29, and GH19, followed by GH20. Among GTS, the major subcategory was observed for GT2 followed by GT4, and GT51. Among CEs, the major subcategory was reported for CE1 followed by CE4, and CE14, whereas in AAs, AA3 showed a higher number followed by AA6, and AA1. Among CBMs, the major subcategory was noted for CBM32 followed by CBM34, and CBM70. Among PLs, the higher subcategory was observed for PL8, and PL4. A total of 2438 CAZyme-encoding sequences were observed among Paenibacillus closely related species tested in the present study. The number of CAZyme-encoding sequences was the highest (n = 491) and the lowest (n = 87) for P. mucilaginosus KNP414 and P. larvae B-3650, respectively (Fig. 8).
Pan genome-core genome analysis
The pangenome analysis was done using nine closely related genomes of non-type strains in the Roary tool. The generated matrix showed the presence/absence profile of genes in selected strains. The most closely related strains of Paenibacillus S-12 belonged to P. alvei A6, P. alvei BLR1 and P. alvei TS-15 (Fig. 9a). Gene presence/absence profiles for P. alvei A6, P. alvei BLR1 and P. alvei TS-15 were similar, while Paenibacillus S-12 displayed a distinct profile from other strains belonging to the same cluster. The pangenome of all nine selected strains contains 17,018 genes, of which the core genome represents 589 genes (18%), the shell genome represent 9162 genes (43%), and the cloud genome represent 7267 genes (38%) (Fig. 9b). On average each strain contained 105 unique genes which correspond to approximately 3% of each genome (Fig. 9c).
Discussion
The S-12 strain was identified as Paenibacillus sp. based on the sequence analysis of the 16 S rRNA gene. The Paenibacillus spp. exhibits environmental survival and increases its population in various ecological niches. However, the evidence at the genomic level is still lacking. A previous report illustrated that bacterial strains with larger genome exhibit more adaptability to complex habitats as larger genomes bear more genes for metabolism and stress tolerance [63]. However, the other studies demonstrated that even a small bacterial genome might also show more competitive, advantages in energy saving, and reproductive efficiency [64, 65]. In the present study, we explore the in-depth genomic analysis to unravel the information about probiotic features, multidrug efflux pumps, transporter genes, presence of antimicrobial, virulence genes, and stress-protectant etc.
In the gut, bile salts exert several deleterious effects like the disruption of the bacterial membrane, denaturation of the proteins, chelation of various ions like iron and calcium, and also modify the eukaryotic gene expression related to the host immunity and defence [66]. To resist these deleterious effects, microorganism evolves defence mechanisms like bile efflux, hydrolysis of bile salts, induction of stress proteins, and reorganization of metabolic pathways [67, 68]. In the test isolate, the various genes encoding bile and sodium symporter, ABC transporter, arginine, and ornithine decarboxylase were noted which might help the bacterium to survive in presence of salts [69, 70]. Bile salts or bile salt hydrolase breaks the complex bile acids and other molecules such as glycine/taurine to allow its diffusion into the cell, leading to increased intracellular acidification [70]. Similarly, arginine and ornithine decarboxylase catalyzes the decarboxylation of arginine/ornithine to putrescine, thereby increasing the intracellular pH [71, 72]. Under these circumstances, F0F1- ATP synthase translocates the protons out and therefore, favours the acid and bile tolerance [73, 74]. It was observed that thepresence of F0F1- ATP synthase and bile-salt symporter minimized the toxic effects of acid and bile effects in probiotic bacteria [69, 70].
The test isolate showed biofilm formation, which can help the bacterium for colonization, and also protection of its host plant against stressors. A previous study showed that biofilm forming plant growth promoting rhizobacterium P. polymyxa colonized the plant roots with the formation of biofilm, and further improved plant resistance to biotic and abiotic stressors [75]. Another study showed that exoglycans producing bacterium P. polymyxa strain 1465 favour the colonization of the bacterium to wheat roots [76].
Most of the Paenibacillus spp. are non-pathogenic, however P. alvei, P. thiaminolyticus and P. sputa showed pathogenicity in terms of respiratory andurinary tract infection, and bacteremia in hemodialysis patients [77,78,79]. The test isolate showed resistance to several antibiotics. The resistance pattern highlights the possible presence of resistant determinants in the S-12. Therefore, we explore the various genomic features in the test isolate through genome sequencing and annotation. The Paenibacillus sp. S-12 genome features many multidrug efflux transporters conferring resistance to many antibiotics. These proteins particularly enhance the efflux, diffusion, and other bacterial defense mechanism against xenobiotics [80, 81]. We noted chloramphenicol acetyltransferase, vancomycin resistance protein vanX, vanH, vanA, & vanW, and vancomycin resistance regulator vanR protein [82]. Additionally, a bacteriocin resistance gene, tetracycline resistance protein TetM, TetO, TetP, TetW, and oxytetracycline resistance protein OtrA were identified. The genome contains some other genes conferring resistance to fluoroquinolones, β-lactams, and dihydrofolate reductase-A inducing resistance to trimethoprim as well as resistance genes to streptothricin [83]. Moreover, the isolate contains the multidrug resistance efflux proteins belonging to the family of RND efflux system, MATE family of antimicrobial extrusion protein, MFS family of multidrug-efflux transporter, and Mex family of multidrug efflux transporteretc. Additionally, the isolate contain the genes inducing resistance to acriflavine, outer membrane multidrug efflux pump, drug transport regulator NfxB, and PmrA multidrug resistance efflux pumps. The present work is the first report showing the detailed characterization of the multidrug efflux system in the Paenibacillus bacterial strain.
The genome of Paenibacillus sp. S-12 features several ABC transporters like oppA (oligopeptide ABC transporter), oppB/C (oligopeptide transport system permease protein), and oppC/D (oligopeptide transport ATP binding protein). In bacteria, the signal recognition particles (SRP) initiate the co-translational protein targeting to the plasma membrane by binding to the N-terminal signal sequence from the translating ribosome. In the strain S-12, we observed the three genes related to SRP such asFtsY (signal recognition particle receptors protein), Ffh (signal recognition particle subunit ffh), SRP-AP (signal recognition particle associated protein), and twin-arginine translocation system TatA/B/C/E (twin-arginine translocation protein systems).
We also identified various metal transporters following annotation of the S-12 genome. In Mg (magnesium)-transporters, various genes like mgtA (Mg++ transport ATPase protein-P), mgtC (Mg++ transport ATPase protein-C), mgtE (Mg-Co-Ni transporters), corA( Mg/Co transport protein), corC (Mg/Co efflux protein), and cat (cation transporting ATPase) were identified. In the copper transporter systems, the genes identified as YcnL (reductase &disulfide isomerise in Cu uptake), YcnK (transcriptional repressors of Cu-uptake), CopA (Cu-translocating P-type ATPase), CopC/D (coper resistance protein), YcnI (membrane protein in Cu uptake), CsoR (repressors of Cu-operon), and CopZ (copper chaperones) [84]. The various genes responsible for nickel (Ni) transport such as NikA (Nickel ABC transporter), NikB/C (Nickel permease protein), NikE (Nickel transport ATP-binding protein), and NikR (Nickel responsive regulator) were identified. Similarly, for cobalt transporters, genes like CbtA/C (cobalt transporter), CbtF (cobalt ABC transporter periplasmic component), and CbtJ/K/L (cobalt ABC transporter) were noted [84]. Besides, genes related to arsenic efflux pumps (arsA, arsB, ACR3) as well as arsenate reductase (arsC, ArrA, ArrB, ArrS) were identified. Among the other metals, we noted the genes conferring resistance to cadmium transport (cadA), cadmium efflux system (cadC), and cadmium resistance protein (cadD). The genes conferring resistance to chromium compounds like chromate resistance protein (ChrI, ChrB), chromate transport protein (ChrA), rhodanese-like protein (ChrE), and superoxide dismutase like protein (ChrF) was identified [84]. Among the Paenibacillus strains, Paenibacillus sp. LYX-1 showed cadmium resistance, and exhibited biocontrol activity [84].
We identified the Type I secretion system that secretes RTX-like adhesion required for auto-aggregation and biofilm formation. The gene like LapB (Type I secretion system ATPase), LapC (membrane fusion protein), LapE (outer membrane component), LapD (membrane bound c-di-GMP receptor), LapP (transglutaminase like cystine protenease), LapL (peptidoglycan associated lipoproteins), and RTX (T1SS-secreted agglutinins) were identified [85]. The presence of flagella, flagellar-associated protein and flagellar regulatory protein helps the bacteria for their colonization and stress survival. Through genome analysis, we identified the flagellin protein (FlaAB), flagellar biosynthesis protein (FlhA/B), flagellar motor protein (MotA/B), and flagellar motor switch protein (FliM/N). Moreover, proteins related to chemotaxis such as CheA/V/Y were also notified. Previous study demonstrated the presence of flagellum in Paenibacillus sp. NAIST15-1, which showed the increased transcription of flagellar genes and hyper-flagellation when transferred from liquid to solid medium [85].
Our genome analysis showed the presence of an NRPS cluster with known predicted functions and a RIPP cluster with unknown products. The discovery of NRPS-lipopeptide highlighted to be attractive pharmaceutical and/ or industrial products. Gene clusters responsible for AMPs, polyketide, polymyxin, and fusaricidin etc., were identified with potent antimicrobial activity [86]. Members of the polyketide group exhibit strong antagonistic activity against food borne pathogens [87]. Presence of these diverse AMPs may increase the ability of Paenibacillus sp. S-12 to fight against pathogenic microorganisms.
The microorganism responsible for plant cell wall degradation plays an essential role inthe recycling of photosynthetically fixed carbon, however, only few microbes are capable to hydrolyze the complex cellulose. Among Paenibacillus genus, P. polymyxa A18 showed higher cellulolytic and hemicellulolytic activities [44]. In this study, we explored the genome of strain S-12 to identify the genes responsible for complex carbohydrate degradation. The carbohydrate enzymes CAZymes are involved in the synthesis as well as the breakdown of complex carbohydrate polymers. The identified CAZymes like GHs and GTs perform the hydrolysis of glycosidic bonds and are commonly noted in Paenibacillus species [88]. The other CAZymes like AAs, PLs, and CBMs are involved in the degradation of several compounds including biopolymers [89]. The improved bioinformatics approach has allowed the identification of gene distribution among its closest relatives by comparative genomics. Gene comparison as well as pengenome exploration leads to discovery of genes involved in strain diversification [90]. The improved genome sequencing approaches pave the way for pangenome investigations in bacteria [91]. The pangenome analysis indicates the S-12 strain harbor many unique genes which are not shared by other strains and thereby gene pool size would increase further increased number of genomes incorporated in the analysis. The open pan-genome indicates that Paenibacillus have the tendency to change its genomic content to adapt to the environment.
Conclusion
Overall, the current findings provide the information about the Paenibacillus sp. S-12 genome that might have acquired or possessed genome features to survive under diverse environmental conditions. The S-12 strain showed probiotic traits essential to thrive through the gastrointestinal transit and also possessed respective genes, making it a strong candidate for probiotics and industrial applications. The presence of antimicrobial genes harnessed by the strain illustrates its ability to mitigate the intestinal pathogens. Moreover, the presence of various plant growth-promoting genes or gene clusters shows its potential to enhance the plant growth and further development of microbial biopesticides.

Phylogenetic tree showing relationship of Paenibacillus sp. S-12 to closely related bacterial strains. The 16 S rRNA gene sequence of closely related species was obtained from NCBI GenBank database. The rooted tree was obtained using Neighbor-joining method of software packages Mega version 7.0, at bootstrap value of (n = 500)

Circular genome map of Paenibacillus sp. S-12 constructed by DNA plotter. Rings from inside represent the following: (1) GC content, (2) GC skew (3) CDS features (4) rRNA (5) tRNA, (6) repeat region, (7) positions labels for genome length (Mbp)

a.Comparison of cluster of orthologs groups in five Paenibacillus species. The analysis was done by using Orthovenn2 using default parameters with protein sequences of Paenibacillus sp. S-12, P. alvei DSM29, P. curdlanolyticus YK9, P. polymyxa ATCC 842, P. polymyxa E681 and P. polymyxa SC2, b. The occurrence table contains groups of gene clusters like cluster count and protein count. Row indicates the orthologous gene cluster for multiple species that summarized as a cell graph and column indicates different closely related bacterial species, C. The pairwise protein sequence comparison for heatmap showing orthologs clusters between S-12 and other closely related strains

The RAST subsystems distribution in the Paenibacillus sp. S-12 genome. The most abundant systems on the category level are shown in the left pie chart, whereas the right column showing the counts of features

a The clusters of orthologus (COGs) analysis in Paenibacillus sp. S-12 genome, b. The metabolic pathway analysis using KEGG Automatic Annotation Server (KAAS) database. KAAS database is used for functional annotation of genes by BLAST comparisons against KEGG-GENES database

The genome of Paenibacillus sp. S-12 was annotated for virulence factor identification using the VFDB database (http://www.mgc.ac.cn/VFs) using the Basic Local Alignment Search Tool (BLASTX) through diamond tool

Identification of putative biosynthetic gene clusters (BGCs) using antiSMASH. antiSMASH analysis identified the 28 BGCs in the Paenibacillus sp. S-12 genome

The distribution of CAZyme domain sequences such as Glycoside hydrolases (GHs); Glycosyl transferases (GTs); Polysaccharide lyases (PLs); Carbohydrate esterases (CEs); Auxiliary activities (AAs); and Carbohydrate-binding modules (CBMs) among the Paenibacillus sp. S-12 and its closely related species

a.The matrix illustrating the presence/absence of genes in selected genome, the clustering of tree is shown on the left side, b. The pie chart shows the proportion of core, shell, and cloud genes c. The gene frequency plot demonstrating the distribution of genes per genome
Data Availability
The datasets generated and/or analysed during the current study are available in the Genbank (https://www.ncbi.nlm.nih.gov/genbank/) under Bioproject and Biosample accession no. PRJNA861075 and SAMN29881974, respectively. The raw illumina data were submitted to the NCBI Sequence Read Archive (SRA) under accession number SRR20556255. The genome sequence was submitted to NCBI and accession no. JASIUF000000000 was assigned.
Abbreviations
- ORF :
-
Open reading frame
- CAZymes :
-
Carbohydrate-active enzyme
- BGCs :
-
Biosynthetic gene clusters
- AFB :
-
American Foulbrood, HGT:Horizontal gene transfer
- AMPs :
-
Antimicrobials proteins
- NRPSs :
-
Nonribosomal peptide synthetases
- PKSs :
-
Polyketide synthase
- RiPPs :
-
Ribosomally synthesized and post-translationally modified peptide
- PBS :
-
Phosphate buffer saline
- IMViC :
-
Indole, Methyl Red, Voges Proskauer
- ZOI :
-
Zone of inhibition
- SEM :
-
Scanning electron microscopy
- TSB :
-
Tryptic soya broth
- CV :
-
Crystal violet
- DSM :
-
Difco sporulation medium
- tRNA :
-
transfer RNA
- rRNA :
-
ribosomal RNA
- COG :
-
Clusters of orthologous groups
- HMM :
-
Hidden Markov Model
- RAST :
-
Rapid annotations using subsytems technology
- MFS :
-
Major facilitator superfamily
- RND :
-
Resistance-nodulation-cell division
References
Ash C, Priest FG, Collins MD. Molecular identification of rRNA group 3 Bacilli (Ash, Farrow,Wallbanks, and Collins) using a PCR probe test. Antonie Van Leeuwenhoek. 1993;64(3):253–60.
Lee FL, Kuo HP, Tai CJ, Yokota A, Lo CC. Paenibacillus taiwanensis sp nov., isolated from soil in Taiwan. Int J Syst Evol Microbiol. 2007;57(6):1351–4.
Mishra AK, Lagier JC, Rivet R, Raoult D, Fournier PE. Non-contiguous finished genome sequence and description of Paenibacillus senegalensis sp nov. Stand Genomic Sci. 2012;7:70–81.
Grady EN, MacDonald J, Liu L, Richman A, Yuan ZC. Current knowledge and perspectives of Paenibacillus: a review. Microb Cell Fact. 2016;15(1):1–18.
Bardají DKR, Furlan JPR, Stehling EG. Isolation of a polyethylene degrading Paenibacillus sp. from a landfill in Brazil. Arch Microbiol. 2019;201(5):699–704.
Tang QY, Yang N, Wang J, Xie YQ, Ren B, Zhou YG, Gu MY, Mao J, Li WJ, Shi YH, Zhang LX. Paenibacillus algorifonticola sp. nov., isolated from a cold spring. Int J Syst Evol Microbiol. 2011;61(9):2167–72.
Gao M, Xie LQ, Wang YX, Chen J, Xu J, Zhang XX, Sui XH, Gao JL, Sun JG. Paenibacillus beijingensis sp. nov., a novel nitrogen-fixing species isolated from jujube garden soil. Antonie Van Leeuwenhoek. 2012;102(4):689–94.
Neuendorf S, Hedtke K, Tangen G, Genersch E. Biochemical characterization of different genotypes of Paenibacillus larvae subsp. larvae, a honey bee bacterial pathogen. Microbiology. 2004;150(Pt 7):2381–90.
Bloemberg GV, Lugtenberg BJ. Molecular basis of plant growth promotion and biocontrol by rhizobacteria. Curr Opin Plant Biol. 2001;4(4):343–50.
Huq MA. Paenibacillus anseongense sp. nov. a silver nanoparticle producing bacterium isolated from rhizospheric soil. Curr Microbiol. 2020;77:2023–30.
Akter S, Wang X, LeeSY, Rahman MM, Park JH, Siddiqi MZ, Balusamy SR, Kihong Nam K, Rahman MS, Huq MA. Paenibacillus roseus sp. nov., a ginsenoside-transforming bacterium isolated from forest soil. Arch Microbiol. 2021;203(7):3997–4004.
Akter S, Huq MA. Biological synthesis of Ginsenoside Rd using Paenibacillus horti sp. nov. isolated from vegetable garden. Curr Microbiol. 2018;75(12):1566–73.
Cochrane SA, Vederas JC. Lipopeptides from Bacillus and Paenibacillus spp.: a gold mine of antibiotic candidates. Med Res Rev. 2016;36:4–31.
Cochrane SA, Findlay B, Bakhtiary A, Acedo JZ, Rodriguez-Lopez EM, Mercier P, et al. Antimicrobial lipopeptide tridecaptin A1 selectively binds to Gram-negative lipid II. Proc Natl Acad Sci USA. 2016;113:11561–6.
Rajesh T, KimYH, Choi YK, Jeon JM, Kim HJ, Park SH, Park HY, Choi KY, Kim H, Kim HJ, Lee SH, Yang YH. Identification and functional characterization of an α-amylase with broad temperature and pH stability from Paenibacillus sp. Appl Biochem Biotechnol. 2013;170(2):359–69.
St John FJ, Rice JD, Preston JF. Paenibacillus sp. strain JDR-2 and XynA (1): a novel system for methylglucuronoxylan utilization. Appl Environ Microbiol. 2006;72:1496–506.
Kim YJ, Sukweenadhi J, Seok JW, Kang CH, Choi ES, Subramaniyam S, Yang DC. Complete genome sequence of Paenibacillus yonginensis DCY84T, a novel plant Symbiont that promotes growth via induced systemic resistance. Stand Genomic Sci. 2017;12(1):1–7.
Naghmouchi K, Baah J, Cudennec B, Drider D. Required characteristics of Paenibacillus polymyxa JB-0501 as potential probiotic. Arch Microbiol. 2013;195:537–43.
Midhun SJ, Neethu S, Vysakh A, Arun D, Radhakrishnan EK, Jyothis M. Antibacterial activity and probiotic characterization of autochthonous Paenibacillus polymyxa isolated from Anabas testudineus (Bloch, 1792). Microb Pathog. 2017;113:403–11.
Li NZ, Xia T, Xu YL, Qiu RR, Xiang H, He D, Peng YY. Genome sequence of Paenibacillus sp. strain Aloe-11, an endophytic bacterium with broad antimicrobial activity and intestinal colonization ability. J Bacteriol. 2012;2117–8.
Hong HA, Duc LH, Cutting SM. The use of bacterial spore formers as probiotics. FEMS Microbiol Rev. 2005;29(4):813–35.
Bentley S. Sequencing the species pan-genome. Nat Rev Microbiol. 2009;7(4):258–9.
GanHM, Hudson AO, Rahman AYA, Chan KG, Savka MA. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics. 2013;14(1):1–4.
Puigbo P, Lobkovsky AE, Kristensen DM, Wolf YI, Koonin EV. Genomes in turmoil: quantification of genome dynamics in prokaryote super genomes. BMC Biol. 2014;12(1):1–19.
Langendries S, Goormachtig S. Paenibacillus polymyxa, a Jack of all trades. Environ Microbiol. 2021. https://doi.org/10.1111/1462-2920.15450.
Keita MB, Padhmanabhan R, Robert C, Delaporte E, Raoult D, Fournier PE, Bittar F. Non-contiguous-finished genome sequence and description of Paenibacillus camerounensis sp. nov. Microb Ecol. 2015;71:990–8.
Sang MK, Kim KD. Biocontrol activity and root colonization by Pseudomonas corrugata strains CCR04 and CCR80 against phytophthora blight of pepper. Biocontrol. 2014;59:437–48.
Sharma RR, Singh D, Singh R. Biological control of postharvest diseases of fruits and vegetables by microbial antagonists: a review. Biol Control. 2009;50:205–21.
Shi Y, Niu KJ, Huang BR, Liu WH, Ma HL. Transcriptional responses of creeping bentgrass to 2,3-butanediol, a bacterial volatile compound (BVC) analogue. Molecules. 2017;22(8):1318.
Jeong H, Choi S-K, Ryu C-M, Park S-H. Chronicle of a soil bacterium: Paenibacillus polymyxa E681 as a tiny guardian of plant and human health. Front Microbiol. 2019;10:467.
Shaheen M, Li J, Ross AC, Vederas JC, Jensen SE. Paenibacillus polymyxa PKB1 produces variants of polymyxin B-type antibiotics. Chem Biol. 2011;18:1640–8.
Lee B, Farag MA, Park HB, Kloepper JW, Lee SH, Ryu CM. Induced resistance by a long-chain bacterial volatile: elicitation of plant systemic defense by a C13 volatile produced by Paenibacillus polymyxa. PLoS ONE. 2012;7(11):e48744.
Furlan JPR, Ralf Lopes R, Stehling EG. Whole–genome sequence–based analysis of the Paenibacillus aquistagni strain DK1, a polyethylene–degrading bacterium isolated from landfill. World J Microbiol Biotechnol. 2021;37(5):1–9.
Yamada Y, Kuzuyama T, Komatsu M, Shin-ya K, Omura S, Cane DE, et al. Terpene synthases are widely distributed in bacteria. Proc Natl Acad Sci USA. 2015;112:857–62.
Zhang LJ, Galo RL. Antimicrobial peptides. Curr Biol. 2016;26:1–21.
Liu Y, Teng K, Wang T, Dong E, Zhang M, Tao Y, Zhong J. Antimicrobial Bacillus velezensis HC6: production of three kinds of lipopeptides and biocontrol potential in maize. J Appl Microbiol. 2019;128:242–25.
Weber T, Blin K, Duddela S, KrugD, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Muller R, Wohlleben W, Breitling R, Takano E, Medema MH. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237–43.
Dickschat JS. Bacterial terpene cyclases. Nat Prod Rep. 2016;33:87–110.
Osbourn A. Secondary metabolic gene clusters: evolutionary toolkits for chemical innovation. Trends Genet. 2010;26(10):449–57.
Palmieri G, Balestrieri M, Proroga YTR, Falcigno L, Facchiano A, Riccio A, Capuano F, Marrone R, Neglia G, Anastasio A. New antimicrobial peptides against foodborne pathogens: from in silico design to experimental evidence. Food Chem. 2016;211:546–54.
Choyam S, Jain PM, Kammara R. Characterization of a potent new-generation antimicrobial peptide of Bacillus. Front Microbiol. 2021;12:710–74.
Rytioja J, Hildén K, Yuzon J, et al. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol Mol Biol Rev. 2014;78:614–49.
Lombard V, Golaconda RH, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–5.
Pasari N, Gupta M, Eqbal D, Yazdani SS. Genome analysis of Paenibacillus polymyxa A18 gives insights into the features associated with its adaptation to the termite gut environment. Sci Rep. 2019;9(1):1–14.
Singh RP, Jha P, Jha PN. The plant-growth-promoting bacterium Klebsiella sp. SBP-8 confers induced systemic tolerance in wheat (Triticum aestivum) under salt stress. J Plant Physiol. 2015;184:57–67.
Eden PA, Schmidt TM, Blakemore RP, Pace NR. Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16S rRNA-specific DNA. Int J Syst Bacteriol. 1991;41(2):324–5.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Hyronimus B, Le Marrec C, Sassi AH, Deschamps A. Acid and bile tolerance of spore-forming lactic acid bacteria. Int J Food Microbiol. 2000;61(2–3):193–7.
Lajhar SA, Brownlie J, Barlow R. Characterization of biofilm forming capacity and resistance to sanitizers of a range of E. coli O26 pathotypes from clinical cases and cattle in Australia. BMC Microbiol. 2018;18(1):41.
Bankevich A, Nurk S, Antipov D, Gurevich A, Dvorkin M, Kulikov AS, Lesin V, Nikolenko S, Pham S, Prjibelski A, Pyshkin A, Sirotkin A, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST server: rapid annotations using Subsystems Technology. BMC Genomics. 2008;9(1):1–15.
Tatusov RL, Natale DA, GarkavtsevLV, Tatusova TA, Shankavaram UT, RaoBS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–8.
Conesa A, GotzS, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization, and analysis in functional genomics research. Bioinformatics. 2005;21(81):3674–6.
Xu L, Ye KX, Dai WH, Sun C, Xu LH, Han BN. Comparative genomic insights into secondary metabolism biosynthetic gene cluster distributions of marine Streptomyces. Mar Drugs. 2019;17:498.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–20.
Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, et al. AntiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47:W81–7.
Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. dbCAN2: a meta server for automated carbohydrate- active enzyme annotation. Nucleic Acids Res. 2018;46(W1):W95–W101.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3.
Hadfield J, Croucher NJ, Goater RJ, Abudahab K, Aanensen DM, Harris SR. Phandango: an interactive viewer for bacterial Population Genomics. Bioinformatics. 2018;34(2):292–3.
Ranea JAG, Buchan DWA, Thornton JM, Orengo CA. Evolution of protein superfamilies and bacterial genome size. J Mol Biol. 2004;336:871–87.
Koskiniemi S, Sun S, Berg OG, Andersson DI. Selection-driven gene loss in bacteria. PLoS Genet. 2012;8(6):e1002787.
Martínez-Cano DJ, Reyes-Prieto M, Martínez-Romero E, Partida-Martínez LP, Latorre A, Moya A, Delaye L. Evolution of small prokaryotic genomes. Front Microbiol. 2015;5:742.
Urdaneta V, Casadesús J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med. 2017;4:163.
Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29(4):625–51.
Ruiz L, Sánchez B, Ruas-Madiedo P, De Los Reyes-Gavilán CG, Margolles A. Cell envelope changes in Bifidobacterium animalis ssp. lactis as a response to bile. FEMS Microbiol Lett. 2007;274(2):316–22.
Khatri I, Sharma G, Subramanian S. Composite genome sequence of Bacillus clausii, a probiotic commercially available as Enterogermina, and insights into its probiotic properties. BMC Microbiol. 2019;19(1):1–15.
Li P, Tian W, Jiang Z, Liang Z, Wu X, Du B. Genomic characterization and probiotic potency of Bacillus sp. DU-106, a highly effective producer of L-lactic acid isolated from fermented yogurt. Front Microbiol. 2018;9:2216.
Barbieri F, Montanari C, Gardini F, Tabanelli G. Biogenic amine production by lactic acid bacteria. Rev Foods. 2019;8(1):17.
Lonvaud-Funel A. Biogenic amines in wines: role of lactic acid bacteria. FEMS Microbiol Lett. 2001;199(1):9–13.
Ruiz L, Margolles A, Sanchez B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front Microbiol. 2013;24(4):396.
Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA. 2014;111:10580–5.
Timmusk S, Grantcharova N, Wagner EGH. Paenibacillus polymyxa invade plant roots and form biofilm. Appl Environ Microbiol. 2005;71:7292–300.
Yogorenkova IV, Tregubova KV, Matora LY, Burygin GL, Ignatov VV. Use of ELISA with anti-exopolysaccharide antibodies to evaluate wheat-root colonization by the rhizobacterium Paenibacillus polymyxa. Curr Microbiol. 2010;61:376–80.
Kim K, Lee K, Yu H, Ryoo S, Park Y, Lee J. Paenibacillus sputi sp. nov., isolated from the sputum of a patient with pulmonary disease. Int J Syst Evol Microbiol. 2010;60(10):2371–6.
Padhi S, Dash M, Sahu R, Panda P. Urinary tract infection due to Paenibacillus alvei in a chronic kidney disease: a rare case report. J Lab Physicians. 2013;5:133.
Ouyang J, Pei Z, Lutwick L, Dalal S, Yang L, Cassai N, Sandhu K, Hanna B, Wieczorek R, Bluth M, Pincus MR. Paenibacillus thiaminolyticus: a new cause of human infection, inducing bacteremia in a patient on hemodialysis. Ann Clin Lab Sci. 2008;38:393–400.
Jack DL, Storms ML, Tchieu JH, Paulsen IT, Saier MH. A broad-specificity multidrug efflux pump requiring a pair of homologous SMR-type proteins. J Bacteriol. 2000;182(8):2311–3.
Saier MH Jr, Paulsen IT Jr. Phylogeny of multidrug transporters. Sem Cell Dev Biol. 2015;12:213.
Galopin S, Cattoir V, Leclercq R. A chromosomal chloramphenicol acetyltransferase determinant from a probiotic strain of Bacillus clausii. FEMS Microbiol Lett. 2009;296(2):185–9.
Eliopoulos GM, Huovinen P. Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis. 2001;32(11):1608–14.
Luo Y, Liao M, Zhang Y, Xu N, Xie X, Fan Q. Cadmium resistance, microbial biosorptive performance and mechanisms of a novel biocontrol bacterium Paenibacillus sp. LYX-1. Environ Sci Pollut Res. 2022;1–15.
Kobayashi K, Kanesaki Y, Yoshikawa H. Surface sensing for Paenibacillus sp. NAIST15-1 flagellar gene expression on solid medium. Appl Environ Microbiol. 2017;83(15):e00585–17.
Lin L, Zheng Q, Wei T, Zhang Z, Zhao C, Zhong H, Xu Q, Lin J, Guo L. Isolation and characterization of fengycins produced by Bacillus amyloliquefaciens JFL21 and its broad-spectrum antimicrobial potential against multidrug-resistant foodborne pathogen. Front Microbiol. 2020;11:579–621.
Chakraborty K, Thilakan B, Raola VK, Joy M. Antibacterial polyketides from Bacillus amyloliquefaciens associated with edible red seaweed Laurenciae papillosa. Food Chem. 2017;218:427–34.
López-Mondéjar R, Zühlke D, Větrovský T, Becher D, Ríedel K, Baldrian P. Decoding the complete arsenal for cellulose and hemicellulose deconstruction in the highly efficient cellulose decomposer Paenibacillus O199. Biotechnol Biofuels. 2016;9(1):104.
Koeck DE, Pechtl A, Zverlov VV, Schwarz WH. Genomics of cellulolytic bacteria. Curr Opin Biotechnol. 2014;29:171–83.
Bottacini F, Morrissey R, Esteban-Torres M, James K, Van Breen J, Dikareva E, Egan M, Lambert J, Van Limpt K, Knol J, O’Connell Motherway M, Van Sinderen D. Comparative genomics and genotype-phenotype associations in Bifdobacterium breve. Sci Rep. 2018;8(1):10633.
Kiu R, Caim S, Alexander S, Pachori P, Hall LJ. Probing genomic aspects of the multi-host pathogen Clostridium perfringens reveals significant pangenome diversity, and a diverse array of virulence factors. Front Microbiol. 2017;8:2485.
Acknowledgements
The author acknowledges the Dept. of Bioengineering and Biotechnology, BIT Mesra for providing the infrastructure.
Funding
The corresponding author acknowledges the Department of Biotechnology, Government of India for providing the Ramalingaswami Re-entry Fellowship. The funding body played no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
RPS supervised the entire study and wrote the original manuscript. KK performed the work pertaining to pertaining to strain isolation and other biochemical characterization. PKS performed the bioinformatics work. Y Ma edited the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animal performed by any of the authors.
Conflict of interest
The author declare no competing interests.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Singh, R.P., Kumari, K., Sharma, P.K. et al. Characterization and in-depth genome analysis of a halotolerant probiotic bacterium Paenibacillus sp. S-12, a multifarious bacterium isolated from Rauvolfia serpentina. BMC Microbiol 23, 192 (2023). https://doi.org/10.1186/s12866-023-02939-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-02939-1