
Abstract
Background
In natural environments, bacteria always live in communities with others where their physiological characteristics are influenced by each other. Bacteria can communicate with one another by using autoinducers. The current knowledge on the effect of quenching bacteria on others is limited to assess the impact of quorum-quenching bacterium Bacillus sp. QSI-1 on proteins pattern and virulence factors production of Aeromonas hydrophila YJ-1. Proteomic analysis was performed to find out protein changes and virulence factors, after 24 h co-culture.
Results
Results showed that several proteins of A. hydrophila YJ-1 were altered, seventy-two differentially expressed protein spots were excised from 2-DE gels and analyzed by MALDI-TOF/TOF MS, resulting in 63 individual proteins being clearly identified from 70 spots. Among these proteins, 50 were divided into 22 classes and mapped onto 18 biological pathways. Mixed-culture growth with Bacillus sp. QSI-1 resulted in an increase of A. hydrophilia proteins involved in RNA polymerase activity, biosynthesis of secondary metabolites, flagellar assembly, and two-component systems. In contrast, mixed culture resulted in a decreased level of proteins involved in thiamine metabolism; valine, leucine and isoleucine biosynthesis; pantothenate and CoA biosynthesis. In addition, the two extracellular virulence factors, proteases and hemolysin, were significantly reduced when A. hydrophila was co-cultured with QSI-1, while only lipase activity was observed to increase.
Conclusions
The information gathered from our experiment showed that Bacillus sp. QSI-1 has a major impact on the expression of proteins, including virulence factors of A. hydrophila.
Similar content being viewed by others
Background
Quorum sensing (QS) is the regulation of gene expression in response to cell density and enables bacteria to regulate the expression of virulence factors and biofilm formation. In nature, bacteria live in mixed populations with other bacterial species. One mechanism used during bacterial species’ competition is QS inactivation, a process commonly referred to as “quorum quenching” (QQ) [1]. During QQ, the QS signal molecules can be inactivated by enzymatic degradation or modification. Such quorum quenching enzymes are wide-spread in the bacterial world and have also been found in eukaryotes. Quorum quenching enzymes can be used to combat bacterial infection [2]. The emergence of antibiotic resistant bacterial strains is a global threat to both animal and human health. The development of new and effective antibiotics is slow, thus therapies that target bacterial QS pathways without killing the bacteria are promising alternatives [3, 4].
Aeromonas hydrophila is an important aquatic pathogen that causes motile aeromonad septicemia in aquatic animals, resulting in great annual economic losses for the aquaculture industry worldwide [5]. Additionally, A. hydrophila is an important zoonotic pathogen that can cause foodborne gastrointestinal and extra-intestinal infections in humans [6]. The pathogenicity of A. hydrophila is closely related to the production of virulence factors and biofilm formation, with virulence factors including serine protease, hemolysin, aerolysin, enterotoxin (cytotoxic enterotoxin) and adhesins, such as fimbriae and S-layer protein [7]. Several studies have indicated that the expression of virulence factors and biofilm formation in A. hydrophila are related to bacterial quorum sensing. A. hydrophila produces N-acyl-L-homoserine lactones such as N-butanoyl homoserine lactone (BHL) and N-hexanoyl homoserine lactone (HHL) as signal molecules [8]. Disruption of QS in A. hydrophila leads to decreased expression of virulence factors [9, 10].
Bacillus spp. live in the environment in the air, soil and water, and some of them have been used as probiotics [11]. Previous studies have shown that Bacillus sp. strain QSI-1 has probiotic properties and can decrease the pathogenicity of A. hydrophila YJ-1 in zebrafish (Danio rerio) and Goldfish (Carassius auratus) models [12,13,14]. We hypothesized that QSI-1 plays important roles in altering the physiological characteristics of A. hydrophila YJ-1, such as metabolism and virulence factor production. In order to improve the understanding of Bacillus sp. QSI-1 effects on A. hydrophila physiology, 2-D gel-based and MALDI-TOF/TOF MS-based proteomic techniques were used to compare global protein expression patterns and extracellular virulence factors of A. hydrophila samples with and without exposure to QSI-1. This study provides new perspectives on bacteria-bacteria interactions. The results of this study likewise suggest that Bacillus sp. QSI-1 can disrupt the virulence of A. hydrophila YJ-1.
Results
Proteome analysis of whole-cell proteins of A. hydrophila YJ-1 co-cultured with Bacillus sp. strain QSI-1
Treatment with probiotics can adjust the gene expression of pathogens, either indirectly via the generation of metabolic molecules or directly via microbe-microbe interactions [15]. To investigate the changes in the A. hydrophila YJ-1 whole-cell protein profiles that occurred in co-culture, 2-D electrophoresis maps using IEF on 24 cm, pH 4–7, nonlinear IPG gels was utilized and compared to whole-cell protein profiles of mono-cultures. The results indicated more than 900 protein spots in a pH range of 4–7 on the Coomassie G-250-stained gels (Fig. 1). Quantitative analysis of the three replicates indicated that 72 protein spots demonstrated more than 2-fold-change difference (P < 0.05) in expression values compared to the mono-culture. The locations of the over-expressed protein spots were marked with numbers. All 72 spots identified in the 2-D electrophoresis gels were excised, digested and analyzed by MALDI-TOF/TOF MS. After this step, 2 protein spots were not identified (spots 6319 and 8534) and pairs of spots correspond to one ID (14 spots represent 7 protein IDs: spots 0058 and 5015; 3717 and 3718; 8341 and 8342; 5619 and 6734; 8628 and 6627; 8718 and 7721; and 9116 and 8124). Of the 63 identified proteins, only 50 could be classified according to KEGG pathways. The changes in protein expression patterns are shown in the supplementary data (Additional file 1: Table S1). Some of the identified proteins were sub-divided into 22 categories based on comparisons with the KEGG database. A number of metabolic pathways and processes were observed to be similar with the identified proteins. These pathways included metabolic pathways, oxidative phosphorylation pathways, pyrimidine metabolism pathways, RNA polymerase-related pathways, purine metabolism pathways, biosynthetic pathways of secondary metabolites, two-component systems, carbohydrate metabolism pathways, and amino acid and nitrogen metabolism pathways. Among these differentially expressed proteins, 24 and 39 were observed to be increased or decreased, respectively, in A. hydrophila YJ-1 co-cultured with QSI-1. To acquire an outline of the elements of differentially communicated proteins that were detected and the potential linkages between them, a Gene ontology (GO) enrichment analysis was performed. In general, 278 proteins were enriched in the biological process (BP) category, with 141 proteins differentially expressed; 32 proteins were marked as cell components (CC), including 5 differentially expressed proteins; a total of 38 differentially expressed proteins were annotated with the molecular function (Fig. 2a). Moreover, the top ten in significantly enriched terms by gene ontology hierarchy (in level 4) were depicted in Fig. 2b. KEGG analysis revealed that most metabolism pathways, including oxidative phosphorylation, pyrimidine metabolism, RNA polymerase activity, and purine metabolism, were significantly improved (Fig. 2c).

Comparative 2-DE maps of proteins extracted from whole cells of A. hydrophila. a: mono-culture, b: co-culture with QSI-1. Proteins were separated by 2-D electrophoresis using pH 4–7 nonlinear IPG strips and 12% SDS-PAGE. The numbers with arrows indicate the identified protein, and the differentially expressed protein IDs are provided in Table S2

Bioinformatic analysis of the identified differentially expressed proteins. a Counts for each category represent the total associated terms in the database with the query protein list. Terms with P-values < 0.05 were statistically significant. b The ten most significantly enriched terms in level 4 gene ontology hierarchy, information of percentage and number of involved proteins in a term are shown in left and right y-axis. c Enriched KEGG pathways are clustered into the metabolism sub-categories; the number of involved proteins in a specific pathway and the corresponding. P-values are shown on the right side of the column
Bacterial growth and extracellular virulence factors of a. hydrophila YJ-1 in mono-culture and in co-culture with QSI-1
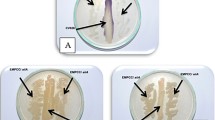
The in vitro growth of the A. hydrophila YJ-1 was assessed in mono-culture and co-cultured with QSI-1 using the plate-count method. Despite the difference in the inoculum, there were no differences in the CFUs from each bacterium after incubation for 24 h at 28 °C (Fig. 3). Disruption of QS in A. hydrophila leads to decreased expression of virulence factors and biofilm formation. Our previous studies have shown that the supernatant of Bacillus sp. strain QSI-1 can inhibit the biofilm formation and virulence factors production [10]. In the present study, we used Transwell plates to investigate the influence of QSI-1 on the production of virulence factors. We evaluated the effect of Bacillus sp. QSI-1 on the production of A. hydrophila YJ-1 QS-controlled virulence. As shown in Table 1, the production of hemolysin and protease was drastically inhibited after co-culturing with QSI-1, whereas the lipase activity increased. The AHLs produced by A. hydrophila YJ-1 when cultured with or without Bacillus sp. QSI-1 was assessed by the production of violacein, as determined by the formation of a zone of purple pigment by Chromobacterium violaceum CV026 (Fig. 4). The results indicated that the QS-associated virulence factors and AHLs produced by A. hydrophila YJ-1 were significantly altered.

Growth curves of A. hydrophila YJ-1 in mono-culture and when co-cultured with QSI-1. The growth rate was measured by the plate counting method. The colony forming units (cfu) were detected from three parallel experiments and are presented as the mean ± standard deviation

Detection of AHLs activity of culture supernatants. Tests shown on the left are from mono-cultures of A. hydrophila, and those on the right are from co-cultures of A. hydrophila and Bacillus sp. QSI-1
Validation of selected altered proteins via qPCR
We selected a subset of the identified differentially expressed proteins for further validation via qPCR. The qPCR results for the selected proteins were consistent with the proteomic results. The mRNA expression level of quorum-regulated virulence genes ser, hem, aer and ahyI, ahyR were all decreased; whereas lip was transcribed at a significantly higher level in the QSI-1 co-culture compared to that in the mono-culture (Fig. 5). The results suggested that the transcriptional levels of all selected genes significantly changed and was consistent with the results of protein levels.

Relative mRNA expression level of selected genes for A. hydrophila YJ-1 co-cultured with Bacillus sp. QSI-1. The averages of the relative fold change of A. hydrophila YJ-1 co-cultured with Bacillus sp. QSI-1 compared to that of A. hydrophila YJ-1 alone are presented. The error bars represent the standard deviation from three different RNA preparations
Discussion
In natural environments, bacteria rarely live alone and are almost exclusively found in communities with other bacteria. When two different bacterial species are placed together in the same environment, there are at least three possible outcomes from this close encounter: neutralism, competition, and antagonism. QS plays an important role in bacterial interactions. QS systems trigger various responses in bacteria, including motility, biofilm formation, extracellular protease production, secondary metabolism and virulence factor gene expression. Therefore, the inhibition of QS by the degradation of AHL molecules has been proposed as a promising alternative to combat bacterial infections. Bacillus sp. QSI-1 can produce quorum-quenching enzymes that can degrade AHLs produced by A. hydrophila. The presence of Bacillus sp. QSI-1 led to a proteomic response in A. hydrophila including the levels of AHLs and QS-regulated extracellular virulence factors. This might be due to the fact that A. hydrophila YJ-1 are exposed to quorum quenching enzymes secreted by QSI-1 in the trans-well system. Our results indicated that the QS-associated virulence factors (hemolysin and protease) and AHLs produced by A. hydrophila YJ-1 were significantly decreased, only lipase activity increased after co-cultured with QSI-1. The high lipase activity under co-culture conditions may be due to the higher importance of lipase for nutritive resources in competition with others [16]. These results suggested that QSI-1 can influence the phenotypes of A. hydrophila because of its production of quorum quenching enzyme. Previous studies have shown that QS plays an important role in the interactions between several species of co-cultured bacteria. The AHL lactonase AiiAAI96 from Bacillus can change the metabolic processes within A. veronii LP-11 cells and inhibit protease production and motility in A. veronii LP-11 [17]. Torabi Delshad et al. [18] showed that co-culture with QQ strains didn’t changed the growth pattern of Yersinia ruckeri as measured by CFU, but the swimming motility, biofilm formation, and the production of virulence factors in Y. ruckeri were decreased. Our results are reversed with those of the lactic acid bacteria, such as Bifidobacterium sp., Enterococcus mundtii and Lactobacillus sp., which are often used for probiotic applications and the inhibition of the growth of pathogens by producing bactericidal factors [19,20,21]. A study by Di Cagno et al. showed that variations in the expression of 58 proteins in Lactobacillus sanfranciscensis DPPMA174, when co-cultured with Lactobacillus plantarum DC400, were affected by interactions with other sourdough Lactobacilli through LuxS-mediated QS mechanisms [22]. Additionally, a study by Canovas et al. (2016) showed an effect of QS cross-talk on the metabolism and virulence of Staphylococcus aureus when co-cultured with Staphylococcus schleiferi in the Galleria mellonella infection model [23]. A study by Rios-Covián et al. provided new insights into the physiological and molecular mechanisms that determine the interactions of Bifidobacterium fragilis and Bifidobacterium longum when grown together [24].
This study provides the first insights into the proteomic response of A. hydrophila when co-cultured with a quorum-quenching bacterium and uncovers possible molecular mechanisms of probiotic bacteria-pathogen interactions. Results showed that most of the proteins are included in more than one category [25]. Proteins involved in different KEGG pathways affected by other species in co-culture system were also found by other studies. In Helicobacter pylori and Streptococcus mitis co-culture system, proteins involved in RNA degradation, nucleotide excision repair, mismatch repair, and lipopolysaccharide biosynthesis were increased in co-cultured H. pylori [6]. While in Bifidobacterium longum and Bifidobacterium breve co-culture system, proteins involved in carbohydrate metabolism, gene regulation, cell envelope biogenesis and transport processes were drastically changed [26].
Conclusion
Our data showed that QSI-1 has a major impact on the expression of proteins, including QS-controlled virulence factors, of A. hydrophila. Our results contribute to the knowledge of competition strategies used by Bacillus sp. during its co-existence with A. hydrophila in natural environment and established new perspectives for the use of Bacillus sp. to control A. hydrophila infection in aquaculture.
Methods
Bacterial strains
The bacterial strains used in this study were A. hydrophila YJ-1 [27], Bacillus sp. strain QSI-1 [10]. A. hydrophila YJ-1 and Bacillus sp. strain QSI-1 were cultured in Luria-Bertani (LB) broth medium (Oxoid, United Kingdom) at 37 °C. For long-term storage, the bacterial strains were preserved at − 70 °C in LB containing 15% (v v− 1) glycerol.
Co-culture experiment
Six-well Costar Transwell plates (Costar, Corning Inc., Kennebunk, ME, USA) with 0.4 μm polyethylene terephthalate (PET) membrane inserts were used to establish a co-culture system that physically separated the two bacterial species but allowed culture medium with secreted bacterial compounds to be exchanged, as previously described by Khosravi et al. [28], with minor modifications. The co-culture assay of A. hydrophila YJ-1 and Bacillus sp. QSI-1 cells that grew for 24 h on LB plates were briefly resuspended in LB to OD600 values of 0.02 (106–107 cfu ml− 1) and 0.008 (105–106 cfu ml− 1), respectively. Next, 0.5 mL aliquots of the Bacillus sp. QSI-1 and A. hydrophila YJ-1 suspensions were added to each well and to the insert well of the 6-well plates, respectively, six-well Costar plates were then used for mono-culture. The cultures were incubated at 28 °C for 24 h, and independent experiments were carried out in triplicate.
Whole-cell protein preparation
Bacterial cells were harvested by centrifugation for 15 min at 4 °C and 12,000 g and then extracted for the whole cell proteins. Whole bacterial cell proteins were prepared as described by Hu et al. and Du et al. [29, 30]. The protein concentration was obtained utilizing the Bradford protein assay (BioRad, CA, USA) with bovine serum albumin (BSA) as a standard according to the manufacturer’s instructions. The samples were stored at − 20 °C until subsequent use.
Two-dimensional gel electrophoresis, in-gel protein digestion, protein identification and database searches
Two-dimensional gel electrophoresis were performed by the method of Sui et al. [31], IEF was performed using the IPG-phor IEF system (BioRad, CA, USA) and Immobiline 24-cm DryStrip IPG strips (pH 4–7). The 2-D electrophoresis was then performed on a 12% SDS-PAGE. Gel electrophoresis was conducted at 16 °C with 1.0 W gel-1 for 1 h and was later alternated with 10 W gel-1 until the dye formed was approximately 1 cm above the bottom of the gel. The protein signals were visualized by staining the gel with Coomassie Brilliant Blue (CBB) G-250. The gel was imaged with a BioRad Fluor-S system and then analyzed using PDQuest (Version 7.2.0; BioRad) software. Protein spots were first identified and later on coordinated automatically in view of the total densities of the gels. For each of the identified spots, the mean relative volume was thought to be equivalent to its expression level at each stage. Identified spots demonstrating more than 2-fold-change difference (P < 0.05) were changed according to the mean relative volume and treated as differentially expressed protein spots. Protein identification by in-gel digestion, mass spectrometric analysis, and database searching using the methods as described in Sui et al. [31].
For GO enrichment analysis UniProt IDs of identified proteins were retrieved from UniProt knowledgebase (UniProtKB) (http://www.uniprot.org/). OmicsBean (http://www.omicsbean.cn), which integrated Gene Ontology (GO) enrichment, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, was employed to analyze the obtained differential abundance proteins (http://www.genome.ad.jp/kegg/).
Detection of extracellular QS-associated virulence factors and enzymatic activities
Twenty-four hours after incubation, the A. hydrophila culture (1 mL) was removed from the well of mono- and co-culture plate, and centrifuged at 6000 g for 5 min. The supernatant was filtered through a 0.45-μm pore-size membrane and stored at − 20 °C until further use.
N-acyl-homoserine lactone (AHL) production was detected by the biosensor Chromobacterium violaceum CV026. Spent supernatants (10 mL) from stationary phase cultures of A. hydrophila YJ-1, from both mono- and co-cultures, were extracted three times with ethyl acetate. The extracts were concentrated to 1 mL, and these concentrated extracts served as exogenous autoinducers and were added to wells inoculated with C. violaceum CV026 to test for violacein production. A result was considered positive for the presence of AHLs when the production of violacein, a purple pigment, by C. violaceum CV026 was observed, and the diameter of purple pigment was detected.
Extracellular enzyme activities were detected by aliquoting 50 μL of supernatant into wells that were cut into LB agar plates supplemented with different substrates, which were then incubated at 37 °C for 24 h. Protease activity was determined by the production of a clear zone of proteolysis around the wells on 1% skim-milk agar plates [32]. The Hemolysin activity was determined by the production of a zone of hemolysis around the holes on blood agar plates containing 2% (v v− 1) sheep blood [33]. The Lipase activity was assayed on Spirit Blue Agar (SBA) (Hi-Media, Mumbai, India) plates, with the presence of lipase visualized as clear blue-colored halos around the wells [26].
Validation of selected altered proteins at the level of mRNA transcription by quantitative real-time PCR
The cultures were grown in triplicate at 28 °C for 24 h under the conditions previously described. Total RNA was extracted using the RNeasy Mini kit (Qiagen Inc., Valencia, CA), the purity and the concentration of RNA was measured with a NanoDrop 2000 spectrophotometer (Thermo Scientific, UK). We examined the expression levels of 8 significantly changed proteins from proteomic results, and virulence factors such as Ser (serine protease), aer (aerolysin), Hem (hemolysin), lip (lipase), and AhyI/R (AHLs synthase and receptor genes). The primers used in this study are shown in Additional file 2: Table S2. Relative gene expression was measured by real-time PCR using the ABI 7500 Fast quantitative PCR system (Applied Biosystems, Carlsbad, USA) and FastStart Universal SYBR Green Master Mix (Rox) (Roche, Mannheim, Germany). 16S rRNA gene was used as a reference gene (forward: 5′-GGGAGTGCCTTCGGGAATCAGA-3′, and reverse: 5′- TCACCGCAACATTCTGATTTG -3′) [15]. Amplifications were performed as described by Feng et al. [34]. Deionized water was used as negative controls. Relative quantification was performed using the 2-ΔΔCt method, where ΔΔCt = (Ct target − Ct 16S rRNA) co-culture − (Ct target − Ct 16S rRNA) mono-culture [35].
Statistical analysis
The data were statistically analyzed using an SPSS software package for Windows version 19.0 (SPSS Inc., Chicago, IL, USA). Differences in the mean values of parameters were tested by oneway analysis of variance and separated by Tukey’s honestly significant difference test (P < 0.05).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- 2-DE:
-
2-dimensional gel electrophoresis
- A. hydrophil :
-
Aeromonas hydrophila
- AHLs:
-
Acylated homoserine lactones
- BHL:
-
N-butanoyl homoserine lactone
- BP:
-
Biological process
- CBB:
-
Coomassie Brilliant Blue
- CC:
-
Cell components
- CFU:
-
Colony-forming unit
- GO:
-
Gene ontology
- HHL:
-
N-hexanoyl homoserine lactone
- IEF:
-
Isoelectric focusing
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LB:
-
Luria-Bertani
- MALDI-TOF/TOF MS:
-
matrix-assisted laser desorption ionization-time of flight mass spectrometry
- mRNA:
-
Messenger ribonucleic acid
- PET:
-
Polyethylene terephthalate
- QQ:
-
Quorum quenching
- QS:
-
Quorum sensing
- RT-qPCR:
-
Reverse transcription quantitative real-time polymerase chain reaction
- SD:
-
Standard deviation
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
References
Uroz S, Dessaux Y, Oger P. Quorum sensing and quorum quenching: the yin and yang of bacterial communication. Chembiochem. 2009;10(2):205–16. https://doi.org/10.1002/cbic.200800521.
Kalia VC. Quorum sensing vs quorum quenching: a battle with no end in sight. New Delhi: Springer; 2015.
Chen F, Gao Y, Chen X, Yu Z, Li X. Quorum quenching enzymes and their application in degrading signal molecules to block quorum sensing-dependent infection. Int J Mol Sci. 2013;14:17477–500. https://doi.org/10.3390/ijms140917477.
Dickey SW, Cheung GYC, Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov. 2017;16(7):457–71. https://doi.org/10.1038/nrd.2017.23.
Beaz-Hidalgo R, Figueras MJ. Aeromonas spp. whole genomes and virulence factors implicated in fish disease. J Fish Dis. 2013;36:371–88. https://doi.org/10.1111/jfd.12025.
Gowda TK, Reddy VR, Devleesschauwer B, Zade NN, Chaudhari SP, Khan WA, et al. Isolation and seroprevalence of Aeromonas spp. among common food animals slaughtered in Nagpu Central India. Foodborne Pathog Dis. 2015;12:626–30. https://doi.org/10.1089/fpd.2014.1922.
Rasmussen-Ivey CR, Figueras MJ, McGarey D, Liles MR. Virulence factors of Aeromonas hydrophila: in the wake of reclassification. Front Microbiol. 2016;25:1337. https://doi.org/10.3389/fmicb.2016.01337.
Swift S, Karlyshev AV, Fish L, Durant EL, Winson MK, Chhabra SR, Williams P, Macintyre S, Stewart GS. Quorum sensing in Aeromonas hydrophila and Aeromonas salmonicida: identification of the LuxRI homologs AhyRI and AsaRI and their cognate N-acylhomoserine lactone signal molecules. J Bacteriol. 1997;179(17):5271–81.
Kozlova EV, Popov VL, Sha J, Foltz SM, Erova TE, Agar SL, et al. Mutation in the S-ribosylhomocysteinase (luxS) gene involved in quorum sensing affects biofilm formation and virulence in a clinical isolate of Aeromonas hydrophila. Microb Pathog. 2008;45:343–54. https://doi.org/10.1016/j.micpath.2008.08.007.
Cui Y, Zhao J, Zhang FR, Guo WP, Chen M, Quan CS, et al. Construction of LuxS gene deletion mutant strain in Aeromonas hydrophila ATCC7966. Chin J Zoo. 2016;32:462–8.
Elshaghabee FMF, Rokana N, Gulhane RD, Sharma C, Panwar H. Bacillus as potential probiotics: status, concerns, and future perspectives. Front Microbiol. 2017;8:1490. https://doi.org/10.3389/fmicb.2017.01490.
Chu WH, Lu F, Zhu W, Kang CT. Isolation and characterization of new potential probiotic bacteria based on quorum-sensing system. J Appl Microbiol. 2011;110:202–8. https://doi.org/10.1111/j.1365-2672.2010.04872x.
Chu WH, Zhou SX, Zhu W, Zhuang XY. Quorum quenching bacteria Bacillus sp. QSI-1 protect zebrafish (Danio rerio) from Aeromonas hydrophila infection. Sci Rep. 2014;4:5446. https://doi.org/10.1038/srep05446.
Zhou SX, Zhang A, Yin H, Chu W. Bacillus sp. QSI-1 modulate quorum sensing signals reduce Aeromonas hydrophila level and alter gut microbial community structure in fish. Front. Cell Infect Microbiol. 2016;6:184. https://doi.org/10.3389/fcimb.2016.00184 eCollection 2016.
O’Toole PW, Cooney JC. Probiotic bacteria influence the composition and function of the intestinal microbiota. Interdiscip Perspect Infect Dis. 2008:175285. https://doi.org/10.1155/2008/175285.
Bourguet N, Goutx M, Ghiglione JF, et al. Lipid biomarkers and bacterial lipase activities as indicators of organic matter and bacterial dynamics in contrasted regimes at the DYFAMED site, NW Mediterranean. Deep Sea Res. II Topical Studies in Oceanography. 2009;56(18):1454–69.
Gui M, Wu R, Liu L, Wang S, Zhang L, Li P. Effects of quorum quenching by AHL lactonase on AHLs, protease, motility and proteome patterns in Aeromonas veronii LP-11. Int J Food Microbiol. 2017;252:61–8. https://doi.org/10.1016/j.ijfoodmicro.2017.04.005.
Torabi Delshad S, Soltanian S, Sharifiyazdi H, Bossier P. Effect of quorum quenching bacteria on growth, virulence factors and biofilm formation of Yersinia ruckeri in vitro and an in vivo evaluation of their probiotic effect in rainbow trout. J Fish Dis. 2018;41(9):1429–38. https://doi.org/10.1111/jfd.12840.
Kailasapathy K, Chin J. Survival and therapeutic potential of probiotic organisms with reference to Lactobacillus acidophilus and Bifidobacterium spp. Immunol Cell Biol. 2000;78:80–8.
Abhisingha M, Dumnil J, Pitaksutheepong C. Selection of potential probiotic Lactobacillus with inhibitory activity against Salmonella and fecal coli form bacteria. Probiotics Antimicrob Proteins. 2017. https://doi.org/10.1007/s12602-017-9304-9308.
Orihuel A, Terán L, Renaut J, Vignolo GM, De Almeida AM, Saavedra ML, Fadda S. Differential proteomic analysis of lactic acid bacteria-Escherichia coli O157:H7 interaction and its contribution to bioprotection strategies in meat. Front Microbiol. 2018;9:1083. https://doi.org/10.3389/fmicb.2018.01083 eCollection 2018.
Di Cagno R, De Angelis M, Calasso M, Vincentini O, Vernocchi P, Ndagijimana M, et al. Quorum sensing in sourdough Lactobacillus plantarum DC400: induction of plantaricin a (PlnA) under co-cultivation with other lactic acid bacteria and effect of PlnA on bacterial and Caco-2cells. Proteomics. 2010;10:2175–90. https://doi.org/10.1002/pmic.200900565.
Canovas J, Baldry M, Bojer MS, et al. Cross-talk between Staphylococcus aureus and other Staphylococcal species via the agr quorum sensing system. Front Microbiol. 2016;7:1733.
Rios-Covián D, Sánchez B, Martínez N, Cuesta I, Hernández-Barranco AM, de Los Reyes-Gavilán CG, et al. A proteomic approach towards understanding the cross talk between Bacteroides fragilis and Bifidobacterium longum in coculture. Can J Microbiol. 2016;62:623–8. https://doi.org/10.1139/cjm-2015-0804.
Rey FA. One protein, many functions. Nature. 2010;468(7325):773–5.
Adnan M, Patel M, Hadi S. Functional and health promoting inherent attributes of Enterococcus hirae F2 as a novel probiotic isolated from the digestive tract of the freshwater fish Catlacatla. PeerJ. 2017;5:e3085. https://doi.org/10.7717/peerj.3085.
Chu WH. Studies on the pathology and control of the bacterial hemorrhage disease in a hybrid crussian carp. Reservoir Fisheries. 2001;21:40.
Khosravi Y, Loke MF, Goh KL, Vadivelu J. Proteomics analysis revealed that crosstalk between Helicobacter pylori and Streptococcus mitis may enhance bacterial survival and reduces carcinogenesis. Front Microbiol. 2016;15:1462.
Hu Q, Ding C, Tu J, Wang X, Han X, Duan Y, Yu S. Immunoproteomics analysis of whole cell bacterial proteins of Riemerella anatipestifer. Vet Microbiol. 2012;157(3–4):428–38. https://doi.org/10.1016/j.vetmic.2012.01.009.
Du D, Gao X, Geng J, Li Q, Li L, Lv Q, Li X. Identification of key proteins and networks related to grain development in wheat (triticum aestivum L.) by comparative transcription and proteomic analysis of allelic variants in TAGW2-6a. Front Plant Sci. 2016;7:922. https://doi.org/10.3389/fpls.2016.00922.
Sui D, Wang B, Shi S, He X. Changes of protein expression during leaves of shrub willow clones in response to salt stress. Acta Physiol Plant. 2015;37(3):51. https://doi.org/10.1007/s11738-015-1811-1.
Takahashi E, Ozaki H, Fujii Y, Kobayashi H, Yamanaka H, Arimoto S, Negishi T, Okamoto K. Properties of hemolysin and protease produced by Aeromonas trota. PLoS One. 2014;9(3):e91149. https://doi.org/10.1371/journal.pone.0091149.
Patel B, Kumari S, Banerjee R, Samanta M, Das S. Disruption of the quorum sensing regulated pathogenic traits of the biofilm-forming fish pathogen Aeromonas hydrophila by tannic acid, a potent quorum quencher. Biofouling. 2017;33(7):580–90. https://doi.org/10.1080/08927014.2017.1336619.
Feng B, Guo Z, Zhang W, Pan Y, Zhao Y. Metabolome response to temperature-induced virulence gene expression in two genotypes of pathogenic Vibrio parahaemolyticus. BMC Microbiol. 2016;16(1):75. https://doi.org/10.1186/s12866-016-0688-5.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT methods. Methods. 2001;25:402–8.
Acknowledgements
The authors are grateful to Tchoudjin Djeukwe Sybille and Robert JC McLean for English editing of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant no. 31672676) and by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for data interpretation and design of the study.
Author information
Authors and Affiliations
Contributions
WC in conceived and supervised the project. SZ performed experimental research work. SZ and ZY drafted the manuscript and performed the statistical analysis. All authors critically reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Functional classes and unclassified proteins of A. hydrophila YJ-1 that were differentially expressed when co-cultured with Bacillus sp. QSI-1. (DOCX 31 kb)
Additional file 2:
Table S2. Primer sequences for qPCR in this study. (DOC 46 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, S., Yu, Z. & Chu, W. Effect of quorum-quenching bacterium Bacillus sp. QSI-1 on protein profiles and extracellular enzymatic activities of Aeromonas hydrophila YJ-1. BMC Microbiol 19, 135 (2019). https://doi.org/10.1186/s12866-019-1515-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1515-6