
Abstract
Background
Basalt is the most common igneous rock on the Earth’s surface covering. Basalt-associated microorganisms drive the cycling and sequestration of different elements such as nitrogen, carbon and other nutrients, which facilitate subsequent pioneer and plant development, impacting long-term regulation of the Earth’s temperature and biosphere. The initial processes of colonization and subsequent rock weathering by microbial communities are still poorly understood and relatively few data are available on the diversity and richness of the communities inhabiting successive and chronological lava flows. In this study, the bacterial communities present on lava deposits from different eruptions of the 1975–84 Krafla Fires (32-, 35- and 39-year old, respectively) at the Krafla, Iceland, were determined.
Results
Three sites were sampled for each deposit (32-, 35- and 39-year old), two proximal sites (at 10 m distance) and one more distant site (at 100 m from the two other sites). The determined chemical composition and metal concentrations were similar for the three basalt deposits. No significant differences were observed in the total number of cells in each flow. 16S rRNA gene amplicon sequencing showed that the most abundant classified phylum across the 3 flows was Proteobacteria, although predominance of Acidobacteria, Actinobacteria and Firmicutes was observed for some sampling sites. In addition, a considerable fraction of the operational taxonomic units remained unclassified. Alpha diversity (Shannon, inverse Simpson and Chao), HOMOVA and AMOVA only showed a significant difference for Shannon between the 32- and 39-year old flow (p < 0.05). Nonmetric multidimensional scaling (NMDS) analysis showed that age significantly (p = 0.026) influenced the leftward movement along NMDS axis 1.
Conclusions
Although NMDS indicated that the (relatively small) age difference of the deposits appeared to impact the bacterial community, this analysis was not consistent with AMOVA and HOMOVA, indicating no significant difference in community structure. The combined results drive us to conclude that the (relatively small) age differences of the deposits do not appear to be the main factor shaping the microbial communities. Probably other factors such as spatial heterogeneity, associated carbon content, exogenous rain precipitations and wind also affect the diversity and dynamics.
Similar content being viewed by others
Background
Igneous rocks constitute about 95% of the Earth’s upper crust and the initial processes of colonization and subsequent rock weathering by microbial communities are still poorly understood. Microorganisms drive the cycling and sequestration of different elements such as nitrogen, carbon and other nutrients, which facilitate subsequent pioneer and plant development, impacting long-term regulation of the Earth’s temperature and biosphere. This interplay between the environment and the biosphere has big implications for the development of the Earth’s atmosphere, not only now, but also for the early Earth [1].
Young unvegetated lava deposits lack readily available nutrients and impose selective conditions on possible colonizing bacterial communities, e.g. nitrogen and carbon need to be supplied via the microorganisms’ N2 and CO2 fixating activity [1,2,3,4]. In addition, exposure to higher UV radiation, temperature fluctuations and desiccation enforce further constraints [5, 6]. Furthermore, since solidified lava cannot retain much water, early microbial colonization depends on precipitation and an exogenous source of nutrients, e.g. atmospheric trace gasses that could also serve as a source of carbon and energy [7, 8]. The best studied igneous rock formations are oceanic basalts, as about 60% of the Earth’s crust consists of basalt, and microorganisms colonizing marine basalts play an important part in the biogeochemical cycling of elements in marine water [9]. In addition, most research for initial colonization of terrestrial rocks has focused on the role of lichens [10], while work on bacteria and their role in volcanic rock weathering was only recently elucidated with Actinobacteria, Proteobacteria and Bacteroidetes being found to be the dominant phyla [1, 2, 5, 11,12,13].
Thus, relatively few data are available on the diversity and richness of the communities inhabiting successive and chronological lava flows. As there is a constant substrate input, chronosequences are formed where older, more weathered substrates are further from the substrate source, which thus serves as a proxy for time and ecosystem development. The latter can be used to study community shifts along this chronosequence to determine how this correlates with habitability development [13]. In this study, we determined and compared the microbial communities of different subsequent lava deposits around the Krafla volcanic field (Iceland) and determined element dynamics along the chronosequence of the lava flows.
Results
Site parameters and lava flow composition
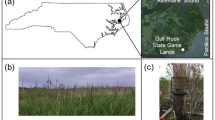
The three different sampling locations represented deposits from different eruptions of the 1975–84 Krafla Fires (32-, 35- and 39-year old, respectively) and showed clear progression from poorly to fully vegetated sites. At each flow, three different points were chosen, resulting in three sites for the three different lava deposits. Two sites were in proximity to each other (10 m distance; designated as year and year*) while a third one (D) was at a distance of 100 m from the two other sites but still within the same lava deposit. Based on the meteoblue climate diagrams (www.meteoblue.com), the mean daily maximum and minimum temperature ranges between − 3° and 12 °C, and − 9 °C and 5 °C, respectively. The mean monthly precipitation ranges between 30 and 84 mm (as rain- and snowfall). The main wind direction is south. The 32-year flow is still exposed to the release of volcanic SO2 and H2S gasses and higher temperatures. No visible vegetation was observed on the 32-year flow. The 35-year flow showed lichen formation while the 39-year old flow already showed the formation of grasses and mosses (Fig. 1). The total number of cells determined by flow cytometry showed that the three sampling sites within the three flows contained between 6.80 × 107 and 7.54 × 108 cells/g (Fig. 2).

Map of the Krafla area in Iceland (Google Earth, Digitalglobe), location and pictures of the three different sampling locations on each of the fissures from the vent and lava flows produced by the eruptions of the 1975–84 Krafla Fires, and meteoblue climate (temperature, precipitation, wind speed and direction- diagrams; www.meteoblue.com)

Total cell number determined by flow cytometry
Basalt composition
The determined chemical composition and metal concentrations were similar for the three flows (Tables 1 and 2).
Taxonomic distribution within each flow and comparison between the different flows and sites
Subsampling was performed based on the lowest amount of reads obtained over the nine different samples, i.e., 32-year sample with a coverage of 3060 reads (Fig. 3). Such a subsampling procedure allowed us to make a fair comparison between the diversity indices for all samples, e.g. avoiding a potential higher diversity due to a higher sequencing coverage.

Rarefaction curve of OTU diversity for each sample
Taxonomic profiling indicated that a large fraction of the operational taxonomic units (OTUs) could not be classified (Fig. 4; Additional file 1: Table S3). At the level of taxonomic families, sites 32, 32* and 32D contained about 35%, 52% and 53% unclassified sequences, respectively. For the 35-year old flow, 35 and 35* contained 41 and 46% unclassified sequences while 35D contained 66% unclassified sequences. The three sites within the 39-year old flow (39, 39* and 39D) contained 40%, 53% and 47% unclassified sequences, respectively. An additional overview of the classified and unclassified number of raw reads, OTUs, single-read OTUs as well as OTUs contributing more than 1% to the taxonomic distribution is given in Table 3. Nevertheless, none of the OTUs identified in the samples were observed in the negative control, which was comprised of sterile water that was subjected to the same processing pipeline as the basalt samples. The major OTUs (relative abundance above 1%) constituted 1 to 18% of all OTUs, the single-read OTUs constituted 0 to 56% of all OTUs. The OTUs shared by the different sampling sites within each flow clearly differed (Fig. 5).

Number of unclassified reads for different taxonomic ranks

Weighted Venn diagram representing the number of OTUs present in the 32-, 35- and 39-year old flow as well as the number of unique and shared OTUs for the sampling sites within one flow
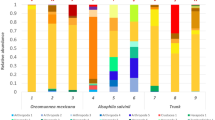
In the 32-year old flow, the most abundant classified phylum across the three sites was Proteobacteria, although site 32 and 32D were predominated by Acidobacteria (mainly Gp4) and Actinobacteria, respectively (Fig. 6). The most abundant OTU belonged to Gp4 (Acidobacteria; OTU9) for site 32 (6.7%) and 32* (4.3%), and was unclassified OTU2 (24.6%) for the more distant site (Fig. 7).

Relative abundance of the bacterial phyla (a) and major bacterial families (b) in the 32-, 35- and 39-year old flows based on 16S rRNA gene sequencing data. Only families with a relative abundance above 2% in at least one sample are shown

The three most abundant OTUs in each sampling site and their respective abundance in the other samples
In the 35-year old flow, the most abundant classified phylum across the three sites also was Proteobacteria, although site 35 was predominated by Acidobacteria (Gp4 family) and site 35D by Actinobacteria (Fig. 6). The most abundant OTU belonged to Gp4 (Acidobacteria; OTU9) for site 35 (7.3%), was unclassified OTU2 for site 35* (11.7%) and unclassified OTU5 for site 35D (9.4%) (Fig. 7).
In the 39-year old flow, the most abundant classified phylum across the three sites was Firmicutes (mainly Clostridiaceae), although site 39* was predominated by Actinobacteria (mainly Geodermatophilaceae) (Fig. 6). The most abundant OTU in site 39 and 39D was OTU5 (37.7% and 44.2%, respectively), and belonged to Geodermatophilaceae for site 39* (OTU10; 16.8%) (Fig. 7).
One hundred and five OTUs were shared between at least one sampling site of the 32-, 35- and 39-year old flow (Additional file 1: Table S3). Three were common to all samples and classified as Clostridium (OTU16), Propionibacterium (OTU22) and Bradyrhizobium (OTU48). Next to the OTUs shared by all three flows, additional OTUs were shared when flows were pairwise compared. The 32- and 35-year old flows additionally shared seven OTUs classified as Acidobacteria (OTU9), Actinobacteria (OTU65, OTU146 and OTU170), Proteobacteria (OTU67 and OTU79) and unclassified (OTU68). The 32- and 39-year old flows additionally shared one unclassified OTU (OTU302). The 35- and 39-year old flows additionally shared six OTUs classified as Bacteroidetes (OTU195), Firmicutes (OTU176), Proteobacteria (OTU113, OTU262 and OTU405) and unclassified (OTU5). The three most abundant OTUs in each sample and their abundance in the other samples indicated the high abundance of a few unclassified OTUs, especially in the 39-year old flow (Fig. 7).
Diversity and NMDS analysis between sites and comparison
Rarefaction curves (Fig. 3) were visualized to indicate if the level of subsampling adequately represented the bacterial diversity in the samples. This showed that subsampled data only fully characterized sampling sites 32D, 35D, 39 and 39D. Therefore, results need to be interpreted cautiously. Nevertheless, alpha diversity (Shannon, inverse Simpson and Chao) calculated on the subsampled and complete data set gave the same conclusion (Table 4, Additional file 2: Table S2). Inverse Simpson and Chao index did not significantly differ between the 32-, 35- and 39-year old flow. Shannon differed significantly between the 32- and 39-year old flow (p < 0.05). Homogeneity of molecular variance (HOMOVA) and analysis of molecular variance (AMOVA) indicated no significant differences between the bacterial community structures of the 32-, 35- and 39-year old flow.
Beta-diversity analysis (Yue & Clayton) indicated that the proximal 32-year old sampling sites as well as the 39 and 39D sampling sites were highly similar (Fig. 8). Likewise, nonmetric multidimensional scaling (NMDS) analysis indicated that the proximal 32-year old sampling sites were similar as well as the 39 and 39D sites, since these ordinated closer together (Fig. 9). The 35-year old sites do not cluster together, possibly indicating that differences for the 35-year flow are more pronounced than for the other flows. Twenty-eight OTUs significantly impacted the downward movement along NMDS axis 2 (with 12 being unclassified). Seventeen OTUs significantly impacted the leftward movement along NMDS axis 2 (with 8 being classified as Proteobacteria). In addition, age significantly (p = 0.026) influenced the leftward movement along NMDS axis 1.

Heat map of the Yue & Clayton measure of dissimilarity

Nonmetric multidimensional scaling (NMDS) analysis of the OTUs of the bacterial communities in the 32-, 35- and 39-year old flows
Discussion
Lava deposits from different eruptions of the 1975–84 Krafla Fires (32-, 35- and 39-year old, respectively) were sampled to determine their bacterial communities. The three different sampling locations, which showed clear progression from poorly to fully vegetated, contained between 6.80 × 107 and 7.54 × 108 cells/g (Fig. 2). Similar cell numbers were found in the rock-soil boundary zones in vegetated sites (both lichens and mosses, and trees) surrounding the Skorradalur lake in Iceland [14]. In contrast, cell number concentrations in recent volcanic deposits studied by Kelly et al. [11] were one log lower.
Taxonomic profiling indicated that a large fraction of the operational taxonomic units (OTUs) could not be classified (Fig. 4; Additional file 1: Table S3). Previously, Lukito et al. [15] also observed that 70% of the OTUs found in Icelandic volcanic glass were unclassifiable. This large fraction of unclassified OTUs could be due to the fact that these environments are not well characterized or because of processing artifacts during DNA amplification as only a low amount of DNA was extracted from the samples (Additional file 3: Table S1). Nevertheless, none of the OTUs identified in the samples were observed in the negative control, which was comprised of sterile water that was subjected to the same processing pipeline as the basalt samples. The low yield could result from natural DNA-binding components, such as humic acids from andosols formation within the older depositions, and DNA sequestration in a hard mineral matrix [15,16,17].
The most abundant phyla (Acidobacteria, Actinobacteria, Firmicutes and Proteobacteria) identified in our samples have already been associated with core basalt communities [3, 18, 19]. Furthermore, Actinobacteria members have been shown to enhance basalt weathering, resist desiccation and possess oligotrophic growth capabilities [5, 14, 20, 21]. They are seen as the key community within the basalt environment as they could possibly act as a C and N source and leach P and S [18, 22]. The different flows also contained Verrucomicrobia (Additional file 1: Table S3), which have been isolated from the Arctic environment and were found to be associated with basalt environments where they greatly contributed to basalt weathering [23]. All flows contained Planctomycetales, Rhizobiales, Rhodospirillales and Sphingomonadales (Additional file 1: Table S3), which have been associated with basalt communities in different volcanic environments [24,25,26,27], deep seafloor sediments and lava formed soil [9, 28]. The heterotrophic capacity of the microbial community on basalt has been proposed to take part in obsidian weathering [29] and could take the available carbon either from the rock (via snowmelt or rain) or through other members of the community [3, 19]. In addition, other community members (found in all three flows) such as those belonging to Chloroflexi, and more specific Ktedonobacter (Additional file 1: Table S3), have also already been associated with the seafloor basalt of Arctic ridges [19] and a volcanic ice cave ecosystem at Mt. Erebus, Antarctica [30]. Other members (found in all three flows) such as Chitinophagaceae (Bacteroidetes) were not yet found to be associated with basalt communities. They probably will stay latent but can flourish when cellulytic substrates substrates become available [31].
Although the Chao index did not significantly differ between the 32-, 35- and 39-year old flow, they were significantly higher than those measured in seafloor basalts (ranging from 12 to 157) [16] as well as in recent Hawaiian volcanic deposits [5]. Also Kelly et al. [11] reported lower numbers for the basaltic Fimmvörðuháls lava flow (Eyjafjallajökull, Iceland) with Chao ranging from 20 to 120 and Shannon index ranging from 1.4 to 2.49. This could indicate that the flows sampled in this study differ from other basaltic environments and are richer in diversity.
Almost no studies have investigated the microbial communities and dynamics present in non-vegetated sections of chronosequences, except for Teixera et al. [32] who showed that in glacier forelands an increase in richness is correlated with increased microbial biomass and activity. Our results indicated that the (relatively small) age difference of the deposits appeared to impact the bacterial community (NMDS); however, this analysis was not consistent with AMOVA and HOMOVA, indicating no significant difference in community structure. The combined results drive us to conclude that the (relatively small) age differences of the deposits do not appear to be the main factor shaping the microbial communities. Probably other factors such as spatial heterogeneity, associated carbon content, exogenous rain precipitations and wind also affect the diversity and dynamics. The impact of spatial heterogeneity can already be seen by comparing the two proximal with the more distant site of the 32-year old flow. In addition, factors such as changes in the mineral substrate, porosity, pH and surface characteristics have also been shown to influence bacterial community composition of terrestrial volcanic rocks [33,34,35]. For instance, rock mineralogy will affect weathering rates, nutrient availability and albedo. The latter influences the temperatures that microbes may experience within the rock [6, 36, 37].
Conclusion
Identification of the bacterial communities present on lava deposits from different eruptions of the 1975–84 Krafla Fires (32-, 35- and 39-year old, respectively), with different vegetation, showed that the (relatively small) age differences of the deposits do not appear to be the main factor shaping the microbial communities.
Methods
Sample sites and sampling
Samples were taken from three different locations on the Leirhnjúkur fissures formed around Krafla, Iceland from the 1975–84 Krafla Fires. During this period, there was significant periodic rifting and faulting along the plate boundary, which was confined to a single system around Krafla. Both rifting episodes included a series of nine small (< 0.2 km3) effusive eruptions that occurred on fissures within the Krafla caldera volcano as well as on the nearby sectors of the fissure swarms [13]. On 27 July 2016, 9 sample sites were defined, three on each of the three lava flows (32-, 35- and 39-year old) originating from the fissure swarms (Fig. 1). The three flows were clearly different in vegetation ranging from almost none (youngest) to fully (oldest) vegetated and were relatively accessible for sampling. Within each flow, three sites were selected. Two sites were in proximity to each other (10 m distance; designated as year and year*) while a third one (D) was at a distance of 100 m from the two other sites but still within the same lava deposit (Fig. 1). Coordinates of the 32-, 35- and 39-year old lava flows are 65°43’39.828’ ’N-16°47’23.3372’ ’W, 65°42’50.5332’ ’N-16°47’26.7576’ ’W and 65°42’56.8872’ ’N-16°47’31.3908’ ’W, respectively.
About 20 g of smaller sized rocks and soil material was sampled at 5 cm below the upper part of the deposits with a sterilized spoon and collected in a 50 ml sterile falcon tube (CELLSTAR® Centrifuge Tubes, Polypropylene, Sterile, Greiner Bio-One, Belgium). These tubes were sealed in three layers of plastic bags and transported at ambient temperature to the laboratory for analysis (see below). The time between collection and analysis was 7 days.
Composition analysis
Five grams of basalt rock material, with a mesh size below 200, were prepared and used to analyze the composition of the different basalt rocks, with lithium metaborate/tetraborate fusion and inductively coupled plasma optical emission spectrometry (ICP-OES)/inductively coupled plasma mass spectrometry (ICP-MS) analysis, and titration for FeO (Actlabs, Canada) (Tables 1 and 2).
Flow cytometry
Five grams of sample was added to 50 ml Butterfield’s Phosphate Buffer (0.25 M KH2PO4 in distilled water, adjusted to pH 7 with 1 M NaOH) and incubated at 30 °C in static conditions. After overnight incubation, part of the buffer solution was analysed with flow cytometry to analyse the total cell numbers (SYBR Green; SG) [38]. Samples were stained according to optimized procedures [39, 40]. Samples were prepared by diluting the sample (to obtain an event rate between 200 and 2000 events/μl) in 0.2 mm filtered mineral water. Next, SG dye (Sigma Aldrich; final concentration of 1×) was added and cell suspensions were incubated at 35 °C for 13 min. Stained bacterial suspensions were analysed on an Accuri C6 (BD, Erembodegem) with a blue (488 nm, 20 mW) and red (640 nm, 14.7 mW) laser, which was calibrated according to the manufacturer’s recommendation. Standard optical filters were used and SG was detected in FL-1 (530/30 nm) with the blue laser. A quality control with 6- and 8-peak fluorescent beads (by manufacturer BD, Erembodegem) and a cleaning cycle were performed prior to experiments to assess both the accuracy (bead count and position) and the cleanliness. Samples were analysed using the Accuri C6 software (version 1.0.264.21).
DNA extraction
To limit DNA extraction bias, two different protocols were used. DNA was extracted from 1 g basalt using the bead-beating cetyl trimethylammonium bromide (CTAB) phenol extraction protocol [41] and the FastPrep protocol [42] without the last Sepharose 4B based purification step [43]. Afterwards, extracted DNA was dissolved in 1xTE buffer and stored at -20 °C.
Illumina sequencing
The DNA concentration was quantified by the Quantifluor dsDNA sample kit on a multi-detection system (Promega, Leiden, the Netherlands). High-throughput amplicon sequencing of the V3-V4 hypervariable region [44] was performed with the Illumina MiSeq platform according to the manufacturer’s guidelines at LGC Genomics GmbH (Berlin, Germany). Sequences were preprocessed using the OCToPUS pipeline combining various preprocessing algorithms [45]. First, both forward and reverse reads are quality checked via looking at k-mer frequency to identify potential false k-mers using the Hammer algorithm implemented in the SPAdes tool. Next, the contigs were created by merging the paired-end reads using the heuristic based on the difference in Phred quality scores of both reads using an updated version of the mothur command “make.contigs” as described in [46]. These contigs were aligned to the SILVA database [47], followed by removing those contigs having an ambiguous base, homopolymer longer than 8 nt, or a length below 400, and those incompliant with the targeted region within the 16S rRNA gene using the mothur commands “align.seqs” and “screen.seqs”, respectively. Next, the aligned sequences were filtered and dereplicated using the mothur commands “filter.seqs” and “unique.seqs”. Secondly, sequencing errors were removed using the IPED algorithm - dedicated to denoise MiSeq amplicon sequencing data [45]. Chimera were detected using the de novo mode of the CATCh algorithm [46]. Creation of the operational taxonomic units (OTUs) was performed using UPARSE ([48] with default parameters (v7.0.1001_i86linux32 – commands sortbysize, cluster_otus, and usearch_global) [42, 45, 46, 49,50,51,52]. The datasets generated and analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) repository (SRP106138).
Statistics
The datasets generated by the two DNA extraction methods were combined for further analyses. Subsampling was performed using the the mothur command “sub.sample” using the amount of reads of the smallest sample. Alpha diversity indices (Chao, Shannon, and inverse Simpson) were calculated using the mothur command “summary.single”. The beta-diversity between the samples was assessed using the Yue & Clayton measure of dissimilarity (mothur command “dist.shared”) and non-metric multidimensional scaling (NMDS) plots (mothur command “nmds”). For the NMDS plot, statistical significant correlation with the coordinates was indicated (p < 0.05) (mothur corr.axes command). The structure of the microbial populations was assessed using the analysis of molecular variance (mothur command “amova”) and homogeneity of molecular variance (mothur command “homova”). Statistical comparison of indices was performed using a one-way ANOVA analysis followed by a post-hoc Tukey test (including multiple testing).
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- CTAB:
-
Cetyl trimethylammonium bromide
- HOMOVA:
-
Homogeneity of molecular variance
- ICP-MS:
-
Inductively coupled plasma mass spectrometry
- ICP-OES:
-
Inductively coupled plasma optical emission spectrometry
- NMDS:
-
Nonmetric multidimensional scaling
- OTU:
-
Operational taxonomic unit
References
Kelly LC, Cockell CS, Herrera-Belaroussi A, Piceno Y, Andersen G, DeSantis T, Brodie E, Thorsteinsson T, Marteinsson V, Poly F. Bacterial diversity of terrestrial crystalline volcanic rocks, Iceland. Microb Ecol. 2011;62(1):69–79.
Kelly LC, Cockell CS, Piceno YM, Andersen GL, Thorsteinsson T, Marteinsson V. Bacterial diversity of weathered terrestrial Icelandic volcanic glasses. Microb Ecol. 2010;60(4):740–52.
Olsson-Francis K, Pearson V, Boardman C, Schofield P, Oliver A, Summers S. A culture-independent and culture-dependent study of the bacteria community from the bedrock soil interface. Adv Microbiol. 2015;5(13):842–57.
Staudigel H, Hart SR. Alteration of basaltic glass: mechanisms and significance for the oceanic crust-seawater budget. Geochim Cosmochim Acta. 1983;47(3):337–50.
Gomez-Alvarez V, King GM, Nüsslein K. Comparative bacterial diversity in recent Hawaiian volcanic deposits of different ages. FEMS Microbiol Ecol. 2007;60(1):60–73.
Mason OU, Di Meo-Savoie CA, Van Nostrand JD, Zhou J, Fisk MR, Giovannoni SJ. Prokaryotic diversity, distribution, and insights into their role in biogeochemical cycling in marine basalts. ISME J. 2009;3(2):231–42.
Chen J, Blume H-P, Beyer L. Weathering of rocks induced by lichen colonization—a review. Catena. 2000;39(2):121–46.
King GM. Contributions of atmospheric CO and hydrogen uptake to microbial dynamics on recent Hawaiian volcanic deposits. Appl Environ Microbiol. 2003;69(7):4067–75.
Cockell CS, Olsson K, Knowles F, Kelly L, Herrera A, Thorsteinsson T, Marteinsson V. Bacteria in weathered basaltic glass, Iceland. Geomicrobiol J. 2009;26(7):491–507.
Fujimura R, Sato Y, Nishizawa T, Nanba K, Oshima K, Hattori M, Kamijo T, Ohta H. Analysis of early bacterial communities on volcanic deposits on the island of Miyake (Miyake-jima), Japan: a 6-year study at a fixed site. Microbes Environ. 2012;27(1):19–29.
Kelly LC, Cockell CS, Thorsteinsson T, Marteinsson V, Stevenson J. Pioneer microbial communities of the Fimmvorethuhals lava flow, Eyjafjallajokull, Iceland. Microb Ecol. 2014;68(3):504–18.
Nemergut DR, Anderson SP, Cleveland CC, Martin AP, Miller AE, Seimon A, Schmidt SK. Microbial community succession in an unvegetated, recently deglaciated soil. Microb Ecol. 2007;53(1):110–22.
Thordarson T, Larsen G. Volcanism in Iceland in historical time: volcano types, eruption styles and eruptive history. J Geodyn. 2007;43(1):118–52.
Summers S, Whiteley AS, Kelly LC, Cockell CS. Land coverage influences the bacterial community composition in the critical zone of a sub-Arctic basaltic environment. FEMS Microbiol Ecol. 2013;86(3):381–93.
Lukito HP, Kouno K, Ando T. Phosphorus requirements of microbial biomass in a regosol and an andosol. Soil Biol Biochem. 1998;30(7):865–72.
Santelli CM, Edgcomb VP, Bach W, Edwards KJ. The diversity and abundance of bacteria inhabiting seafloor lavas positively correlate with rock alteration. Environ Microbiol. 2009;11(1):86–98.
Herrera A, Cockell CS. Exploring microbial diversity in volcanic environments: a review of methods in DNA extraction. J Microbiol Methods. 2007;70(1):1–12.
Cockell CS, Kelly LC, Marteinsson V. Actinobacteria—an ancient phylum active in volcanic rock weathering. Geomicrobiol J. 2013;30(8):706–20.
Lysnes K, Thorseth IH, Steinsbu BO, Øvreås L, Torsvik T, Pedersen RB. Microbial community diversity in seafloor basalt from the Arctic spreading ridges. FEMS Microbiol Ecol. 2004;50(3):213–30.
Lechevalier HA, Lechevalier MP. Biology of actinomycetes. Annu Rev Microbiol. 1967;21(1):71–100.
Bergen B, Herlemann DP, Labrenz M, Jürgens K. Distribution of the verrucomicrobial clade Spartobacteria along a salinity gradient in the Baltic Sea. Environ Microbiol Rep. 2014;6(6):625–30.
Paul D, Kumbhare SV, Mhatre SS, Chowdhury SP, Shetty SA, Marathe NP, Bhute S, Shouche YS. Exploration of microbial diversity and community structure of Lonar Lake: the only hypersaline meteorite crater Lake within basalt rock. Front Microbiol. 2016;6:1553. https://doi.org/10.3389/fmicb.2015.01553.
Henri PA, Rommevaux-Jestin C, Lesongeur F, Mumford A, Emerson D, Godfroy A, Ménez B. Structural Iron (II) of basaltic glass as an energy source for Zetaproteobacteria in an abyssal plain environment, off the mid Atlantic ridge. Front Microbiol. 1518;6:2016.
Kim JS, Lee KC, Kim DS, Ko SH, Jung MY, Rhee SK, Lee JS. Pyrosequencing analysis of a bacterial community associated with lava-formed soil from the Gotjawal forest in Jeju, Korea. MicrobiologyOpen. 2015;4(2):301–12.
Einen J, Kruber C, Øvreås L, Thorseth I, Torsvik T. Microbial colonization and alteration of basaltic glass. Biogeosci Discuss. 2006;3(2):273–307.
Oh T-J, Han S-R, Kang S, Park H, Kim AY. Complete genome sequence of the xylan-degrading Mucilaginibacter sp. strain PAMC26640 isolated from an Arctic lichen. J Biotechnol. 2016;227:23–4.
Erlacher A, Cernava T, Cardinale M, Soh J, Sensen CW, Grube M, Berg G. Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front Microbiol. 2015;6:53.
Walsh J, Clarke E. The role of fractional crystallization in the formation of granitic and intermediate rocks of the Beinn Chaisgidle Centre, Mull, Scotland. Mineral Mag. 1982;45(337):247–55.
Herrera A, Cockell CS, Self S, Blaxter M, Reitner J, Arp G, Drose W, Thorsteinsson T, Tindle AG. Bacterial colonization and weathering of terrestrial obsidian in Iceland. Geomicrobiol J. 2008;25(1):25–37.
Tebo BM, Davis RE, Anitori RP, Connell LB, Schiffman P, Staudigel H. Microbial communities in dark oligotrophic volcanic ice cave ecosystems of Mt. Erebus, Antarctica. Front Microbiol. 2015;6:179. https://doi.org/10.3389/fmicb.2015.00179.
Carson JK, Campbell L, Rooney D, Clipson N, Gleeson DB. Minerals in soil select distinct bacterial communities in their microhabitats. FEMS Microbiol Ecol. 2009;67(3):381–8.
Teixeira LC, Peixoto RS, Cury JC, Sul WJ, Pellizari VH, Tiedje J, Rosado AS. Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J. 2010;4(8):989–1001.
Barker W, Welch S, Chu S, Banfield J. Experimental observations of the effects of bacteria on aluminosilicate weathering. Am Mineral. 1998;83(11):1551–63.
Bennett P, Rogers J, Choi W, Hiebert F. Silicates, silicate weathering, and microbial ecology. Geomicrobiol J. 2001;18(1):3–19.
Hall K, Lindgren BS, Jackson P. Rock albedo and monitoring of thermal conditions in respect of weathering: some expected and some unexpected results. Earth Surf Process Landf. 2005;30(7):801–12.
Wolff-Boenisch D, Gislason SR, Oelkers EH, Putnis CV. The dissolution rates of natural glasses as a function of their composition at pH 4 and 10.6, and temperatures from 25 to 74 C. Geochim Cosmochim Acta. 2004;68(23):4843–58.
Edwards KJ, Bach W, McCollom TM, Rogers DR. Neutrophilic iron-oxidizing bacteria in the ocean: their habitats, diversity, and roles in mineral deposition, rock alteration, and biomass production in the deep-sea. Geomicrobiol J. 2004;21(6):393–404.
Berney M, Hammes F, Bosshard F, Weilenmann HU, Egli T. Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight kit in combination with flow cytometry. Appl Environ Microbiol. 2007;73(10):3283–90.
Buysschaert B, Byloos B, Leys N, Van Houdt R, Boon N. Reevaluating multicolor flow cytometry to assess microbial viability. Appl Microbiol Biotechnol. 2016;100(21):9037–51.
Van Nevel S, Koetzsch S, Weilenmann HU, Boon N, Hammes F. Routine bacterial analysis with automated flow cytometry. J Microbiol Methods. 2013;94(2):73–6.
Griffiths RI, Whiteley AS, O'Donnell AG, Bailey MJ. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol. 2000;66(12):5488–91.
Vilchez-Vargas R, Geffers R, Suárez-Diez M, Conte I, Waliczek A, Kaser VS, Kralova M, Junca H, Pieper DH. Analysis of the microbial gene landscape and transcriptome for aromatic pollutants and alkane degradation using a novel internally calibrated microarray system. Environ Microbiol. 2013;15(4):1016–39.
Mysara M, Njima M, Leys N, Raes J, Monsieurs P. From reads to operational taxonomic units: an ensemble processing pipeline for MiSeq amplicon sequencing data. Gigascience. 2017;6(2):1–10.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–20.
Mysara M, Leys N, Raes J, Monsieurs P. IPED: a highly efficient denoising tool for Illumina MiSeq paired-end 16S rRNA gene amplicon sequencing data. BMC Bioinformatics. 2016;17(1):192.
Mysara M, Saeys Y, Leys N, Raes J, Monsieurs P. CATCh, an ensemble classifier for chimera detection in 16S rRNA sequencing studies. Appl Environ Microbiol. 2015;81(5):1573–84.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–6.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35(21):7188–96.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Jost L. Entropy and diversity. Oikos. 2006;113(2):363–75.
Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005;71(3):1501–6.
Acknowledgements
We thank Kai Finster and Charles Cockell for the coordination of the BIOROCK project. We thank the European Space Agency (ESA) and the Belgian Federal Science Policy Office (BELSPO) for their support in the framework of the PRODEX Programme. We would also like to thank the organizers of the summer school “Biosignatures and the Search For Life on Mars”, co-organized by the Nordic Network of Astrobiology, the European Astrobiology Campus, and the EU COST Action “Origins and Evolution of Life on Earth and in the Universe”.
Funding
This work was supported by the European Space Agency (ESA-PRODEX), Belgian Science Policy (BELSPO) through the E-GEM/BIOROCK project (Bo Byloos) and the Inter-University Attraction Pole (IUAP) “μ-manager” funded by the Belgian Science Policy (BE, 305 P7/25). Participation to the EU COST Action “Origins and Evolution of Life on Earth and in the Universe” summer school was supported by EU COST Action TD1308. The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing this manuscript.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the NCBI Sequence Read Archive (SRA) repository (SRP106138), https://www.ncbi.nlm.nih.gov/sra/. In addition, samples were taken during, and according to the guidelines of the summer school “Biosignatures and the Search For Life on Mars”, co-organised by the Nordic Network of Astrobiology, the European Astrobiology Campus and the EU COST Action “Origins and Evolution of Life on Earth and in the Universe”.
Author information
Authors and Affiliations
Contributions
BB provided experimental data. PM and MM performed the bioinformatics analyses. BB and RVH wrote the manuscript, and analysed and interpreted the results. RVH, NL and NB contributed to conception and design. All authors have read and approved the manuscript, and ensure that this is the case.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S3. Overview of the subsampled dataset generated in this study. (XLSX 109 kb)
Additional file 2:
Table S2. Diversity indexes for each of the sampled sites calculated for the complete and subsampled data. (DOCX 20 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Byloos, B., Monsieurs, P., Mysara, M. et al. Characterization of the bacterial communities on recent Icelandic volcanic deposits of different ages. BMC Microbiol 18, 122 (2018). https://doi.org/10.1186/s12866-018-1262-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-018-1262-0