
Abstract
Background
The sulfate-reducing bacterium Desulfatiglans anilini can grow with phenol as sole source of carbon and energy under strictly anaerobic, sulfate-reducing conditions. In the nitrate-reducing bacterium Thauera aromatica, the enzymes involved in phenol degradation have been well elucidated, whereas the anaerobic phenol degradation pathway by D. anilini was not studied in detail yet.
Results
The pathway of anaerobic phenol degradation by the sulfate-reducing bacterium Desulfatiglans anilini was studied by identification of genes coding for phenylphosphate synthase (encoded by pps genes) and phenylphosphate carboxylase (encoded by ppc genes) in the genome of D. anilini, by analysis of the transcription and translation of pps-ppc genes, and by measurement of phenylphosphate synthase activity in cell-free extracts of phenol-grown cells. The majority of genes involved in phenol degradation were found to be organized in one gene cluster. The gene cluster contained genes ppsα (phenylphosphate synthase alpha subunit), ppsβ (phenylphosphate synthase beta subunit), ppcβ (phenylphosphate carboxylase beta subunit), as well as 4-hydroxybenzoyl-CoA ligase and 4-hydroxylbenzoyl-CoA reductase-encoding genes. The genes ppsγ (phenylphosphate synthase gamma subunit), ppcα (phenylphosphate carboxylase alpha subunit) and ppcδ (phenylphosphate carboxylase delta subunit) were located elsewhere in the genome of D. anilini, and no obvious homologue of ppcγ (phenylphosphate carboxylase gamma subunit) was found in the genome. Induction of genes pps and ppc during growth on phenol was confirmed by reverse transcription polymerase chain reaction. Total proteome analysis revealed that the abundance of enzymes encoded by the gene cluster under study was much higher in phenol-grown cells than that in benzoate-grown cells. In in-vitro enzyme assays with cell-free extracts of phenol-grown cells, phenylphosphate was formed from phenol in the presence of ATP, Mg2+, Mn2+, K+ as co-factors.
Conclusions
The genes coding for enzymes involved in the anaerobic phenol degradation pathway were identified in the sulfate-reducing bacterium D. anilini. The results indicate that the first steps of anaerobic phenol degradation in D. anilini are phosphorylation of phenol to phenylphosphate by phenylphosphate synthase and carboxylation of phenylphosphate by phenylphosphate carboxylase.
Similar content being viewed by others

Background
Phenol is an important industrial commodity used as a precursor for the production of plastics, polycarbonates, epoxies, detergents and pharmaceutical drugs. Its wide use and toxicity has caused serious contaminations of waters and soils. Biological phenol degradation is an economic and effective method to deal with these contaminants without causing secondary pollution problems [1].
Anaerobic phenol degradation has been studied in detail with the denitrifying bacterium Thauera aromatica. Initially, phenol is phosphorylated to phenylphosphate [2], which is subsequently carboxylated to 4-hydroxybenzoate [3, 4]. The enzyme involved in the first reaction is phenylphosphate synthase (encoded by pps genes), which converts phenol and ATP to phenylphosphate, AMP, and phosphate [5]. Phenylphosphate synthase contains three subunits (α, β, γ), and their encoding genes are located adjacent to each other in one operon (Fig. 1). The α-subunit (70 kDa) containing a conserved histidine residue alone can catalyze the exchange of free [14C] phenol and the phenol moiety of phenylphosphate, but not the phosphorylation of phenol. The β-subunit (40 kDa) is required in the phosphorylation of phenol, which can transfer a diphosphoryl group to the conserved histidine residue in the α-subunit [6]. The reaction is stimulated by the addition of γ-subunit (24 kDa), but the exact function of the γ-subunit is unknown [5].

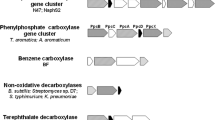
Organization of genes encoding phenylphosphate synthase (ppsαβγ) and phenylphosphate carboxylase (ppcαβγδ) in T. aromatica and the proposed anaerobic phenol degradation gene cluster in D. anilini. The directions of transcription are indicated by arrows. Similar ORFs are presented by the same shades and patterns
The enzyme involved in the subsequent carboxylation of phenylphosphate with CO2 to 4-hydroxybenzoate is phenylphosphate carboxylase (encoded by ppc genes), which consists of four subunits (α, β, γ, δ) [3, 4, 7]. The exchange of 14CO2 and the carboxyl group of 4-hydroxybenzoate was catalyzed by three of the subunits (α, β, γ; 54, 53, and 10 KDa). Phenylphosphate carboxylation was restored when the δ subunit (18 KDa) was added [7]. The δ subunit is assigned to the hydratase/phosphatase protein family and can catalyze alone a very slow hydrolysis of phenylphosphate. The genes coding for these four subunits are located adjacent to each other in one operon (Fig. 1). K+ and divalent metal cations (Mg2+ or Mn2+) are required for phenylphosphate carboxylase activity, and oxygen is an inhibitor for phenylphosphate carboxylase activity. 4-hydroxybenzoate is then catalyzed by 4-hydroxybenzoate CoA ligase to 4-hydroxybenzoyl CoA [8], which is converted to the central intermediate benzoyl-CoA by 4-hydroxybenozyl-CoA reductase [9].
The same phenol degradation pathway was proposed earlier for the iron-reducing bacterium Geobacter metallireducens GS-15 [10]. The initial steps of phenol degradation in G. metallireducens are accomplished by phenylphosphate synthase and phenylphosphate carboxylase as known from Thauera aromatica. The phenol induced gene cluster (pps-ppc) was identified in the genome of G. metallireducens, which revealed some differences compared to the corresponding gene cluster in T. aromatica: it is not induced specifically in phenol-grown cells and it only contains a ppc β homologue. In the fermenting bacterium Sedimentibacter hydroxybenzoicus, phenol is most likely carboxylated by an ATP-dependent 4-hydroxybenzoate decarboxylase [11, 12]. In a newly isolated strain of the sulfate-reducing bacterium Desulfatiglans anilini, the phenol degradation pathway appears to be the same as in Thauera aromatica [13]. Yet, phenol degradation was not studied at the biochemical and proteome level before in sulfate reducing bacteria. In the present study, we identified the catabolic enzymes and their genes involved in anaerobic degradation of phenol in the sulfate reducing bacterium Desulfatiglans anilini.
Results
Anaerobic growth with phenol or benzoate
The growth of Desulfatiglans anilini on phenol or benzoate was investigated. 2 mM phenol or 2 mM benzoate was supplied to D. anilini cultures as the only source of electrons with Na2SO4 as the electron acceptor (Fig. 2). The doubling time of D. anilini on benzoate is 4.4 days, which is slightly shorter than that on phenol (6.6 days). Culture samples were taken at different time points, and metabolites were analyzed by HPLC. No intermediate organic degradation products were detected in the growth medium. 2 mM phenol or benzoate was consumed in around 20 days along with approximately 8 mM Na2SO4 being reduced. Cells of late logarithmic phase cultures that had been pre-grown for 10 generations on the respective substrates were harvested and used for the following experiments.

Anaerobic growth of D. anilini with 2 mM phenol or 2 mM benzoate plus sulfate (10 mM) as electron acceptor
Similarity search of genes involved in phenol metabolism
The enzymes reported to be involved in phenol degradation by T. aromatica (ppsα, ppsβ, ppsγ, ppcα, ppcβ, ppcγ, ppcδ, ORF7–11 and ORF13–14) [14, 15] or G. metallireducens (BamQ, BamA, BamR and BamB) were used for a similarity search in the genome of D. anilini by IMG/M blast or NCBI blast. As shown in Table 1, the proteins with locus tags H567DRAFT_02052, H567DRAFT_02051, H567DRAFT_02059, H567DRAFT_02049 and H567DRAFT_02056 displayed the highest identities of 38–56% to ppsα, ppsβ, ppcβ, ORF14 and ORF8 of T. aromatica individually. The genes coding for the above-mentioned proteins are located in one gene cluster, indicating that this gene cluster could be involved in phenol degradation. However, the proteins that showed highest similarities to the genes ppsγ, ppcδ and ppcα in T. aromatica were the acetoin utilization protein AcuB (locus tag H567DRAFT_03126), KDO 8-P phosphatase (locus tag H567DRAFT_00862) and phenylphosphate carboxylase beta subunit (locus tag H567DRAFT_03563) individually, whose encoding genes are not located in the same gene cluster. Genome analysis did not reveal obvious homologues of ppcγ. For the ORFs 7, 9, 10, 11, 13, the genes showing high similarities were not localized in this gene cluster.
For the downstream metabolism of phenol, the degradation pathway of benzoyl-CoA consists of benzoyl-CoA reductase (BamB-I) [16, 17], a cyclohexadienoyl-CoA hydratase (BamR) [18], a hydroxyenoyl-CoA dehydrogenase (BamQ) [16], and an oxoenoyl-CoA hydrolase (BamA) [19]. The genes coding for proteins which display the highest similarities to BamQ, BamA and BamR of G. metallireducens are located adjacent to each other in the genome of D. anilini.
Reverse transcription PCR analysis
Reverse transcription polymerase chain reaction (RT-PCR) experiments were performed with mRNA extracted from cells grown on phenol or benzoate (Fig. 3) to test whether the putative genes of ppsα, ppsβ, ppsγ, ppcα, ppcβ, and ppcδ in the genome of D. anilini were induced by phenol. With cDNA from phenol-grown cells, all genes under study were amplified. With cDNA from benzoate-grown cells, no amplifications of ppsβ, ppcα and ppcδ were obtained. This result indicates that transcription of all genes (ppsα, ppsβ, ppsγ, ppcα, ppcβ, and ppcδ) was induced in phenol-grown cells, but not all of them were induced in benzoate-grown cells. Hence, this result confirmed the involvement of these genes in phenol degradation by D. anilini. In a control PCR experiment, genomic DNA instead of cDNA from mRNA was used as the template, and DNA fragments of the expected sizes were obtained in all cases (Additional file 1: Figure S1). The gene coding for dissimilatory adenylylsulfate reductase alpha subunit precursor was transcribed in both phenol-grown and benzoate-grown cells and used as the house-keeping gene reference.

Reverse transcriptase PCR of genes ppsα, ppsβ, ppsγ, ppcα, ppcβ and ppcδ in D. anilini. (A) Reverse transcriptase PCR products of ppsα, ppsβ, ppsγ, ppcα, ppcβ, ppcδ and AsrA (dissimilatory adenylylsulfate reductase alpha subunit precursor; housekeeping gene) using the cDNA from phenol-grown cells as template. (B) Reverse transcriptase PCR products of ppsα, ppsβ, ppsγ, ppcα, ppcβ, ppcδ and AsrA (dissimilatory adenylylsulfate reductase alpha subunit precursor; housekeeping gene) using the cDNA from benzoate-grown cells as template
Total proteomics analysis
To further verify the expression of putative genes involved in phenol degradation by D. anilini, the proteome of phenol-grown cells was compared to that of benzoate-grown cells by total proteomics analysis. Equal amounts of protein from phenol-grown cells or benzoate-grown cells were analyzed, and all protein abundances were quantified by label-free protein quantification (LFQ) (Fig. 4). The LFQ of enzymes encoded by the putative phenol degradation gene cluster (locus tag from H567DRAFT_02049 to H567DRAFT_02059) in phenol-grown cells were much higher than that in benzoate-grown cells (Fig. 4a). The gene cluster under study is a 14.4 kb gene cluster in the genome of D. anilini. The putative gene ppcα (locus tag H567DRAFT_03563) which is not located in this gene cluster was specifically induced in phenol-grown cells. Nevertheless, the putative gene ppsγ (locus tag H567DRAFT_03126) did not exhibit higher abundance in phenol-grown cells than in benzoate-grown cells. These results further verified the participation of the gene cluster in phenol degradation. Hence, phenol is presumed to be degraded through a similar pathway as in T. aromatica.

Total proteome analysis of cell-free extracts from the cells of D. anilini grown on phenol (black bars) and benzoate (grey bars). (A) label-free quantification intensity (LFQ-intensity) of the enzymes encoded by pps-ppc gene cluster (locus tag 02049–02059) and putative ppsγ (locus tag 03126) and ppcα (locus tag 03563) in D. anilini grown with phenol or benzoate. (B) LFQ-intensity of enzymes (6-hydroxycyclohex-1-ene-1-carboxyl-CoA dehydrogenase, 6-ketocyclohex-1-ene-1-carbonyl-CoA hydrolase, cyclohexa-1, 5-diene-1-carbonyl-CoA hydratase, tungsten-dependent benzoyl-CoA reductase subunit bamB) involved in benzoyl CoA metabolism in D. anilini grown with phenol or benzoate. The LFQ intensity value of the enzymes from phenol-grown cells are labeled at the end of the columns. The lowest LFQ intensity value observed in total proteomics analysis was 109,120, and the highest LFQ intensity observed is 16,493,000,000
The total proteome data of phenol-grown cells and benzoate-grown cells also demonstrate the existence of the putative enzymes to be involved in anaerobic benzoyl-CoA degradation. The label-free quantification (LFQ) intensity of these enzymes (H567DRAFT_01120 6-hydroxycyclohex-1-ene-1-carboxyl-CoA dehydrogenase, H567DRAFT_01121 6-ketocyclohex-1-ene-1-carbonyl-CoA hydrolase, H567DRAFT_01122 cyclohexa-1, 5-diene-1-carbonyl-CoA hydratase) did not show significant differences between phenol-grown cells or benzoate –grown cells (Fig. 4b).
Phenylphosphate synthase activity
In-vitro enzyme activity assays were carried out with cell-free extracts of phenol- or benzoate-grown cells. The enzyme activity of phenylphosphate synthase was observed by detecting the formation of phenylphosphate over time, using phenol and ATP as the substrates with cell-free extracts of phenol-grown cells. Fig. 5 shows that these cell-free extracts were able to convert phenol to phenylphosphate with ATP as a co-substrate and Mg2+, Mn2+, and K+ as cofactors. Extracts of phenol-grown T. aromatica cells catalyzed the MgATP-dependent formation of [14C] phenylphosphate from [U-14C] phenol at a specific rate of 1.5 nmol min− 1 mg− 1 of protein [5]. In our study, the rate of phenylphosphate formation catalyzed by phenylphosphate synthase was tested as 0.52 nmol min− 1 mg− 1 of protein which is lower than that of T. aromatica cells. The activity was inhibited by oxygen, and no activity was measured with cell free extracts of benzoate grown cells.

Time course of enzymatic phenylphosphate formation from phenol and ATP by the putative phenylphosphate synthase discontinuously assayed by HPLC with crude cell-free extract as sample
Phenylphosphate carboxylase activity
The activity of phenylphosphate carboxylase was assayed either by spectrophotometric assays or HPLC [3]. In spectrophotometric assays no change of absorption could be measured as a consequence of phenylphosphate consumption at 235 nm or 4-hydroxybenzoate production at 280 nm. No substrate consumption or product formation was observed by HPLC, neither with CO2 nor with CO as co-substrate.
Discussion
In the present study, the initial steps of phenol activation in a sulfate-reducing bacterium were studied by characterizing the transcription and expression of pps- and ppc- like ORFs and in-vitro phenylphosphate synthase assays. The results revealed that the phenol degradation pathway in this sulfate-reducing bacterium D. anilini (Fig. 6) is analogous to the known phenol degradation pathway in the nitrate-reducing bacterium T. aromatica.

Proposed pathway of anaerobic phenol degradation by D. anilini. Numbers in dark ellipses indicate the locus tags of the enzymes
The first step for phenol activation in D. anilini is a phosphorylation of phenol to phenylphosphate catalyzed by phenylphosphate synthase. Homologues of the three genes coding for the three subunits of phenylphosphate synthase (ppsαβγ) in T. aromatica are present in the genome of D. anilini. The genes ppsα (locus tag H567DRAFT_02052) and ppsβ (locus tag H567DRAFT_02051) are two adjacent genes showing homologies to pyruvate, water dikinase, and are transcribed and translated during growth on phenol. The putative gene ppsγ (locus tag H567DRAFT_03126) was transcribed both in phenol- and in benzoate-grown cells. The expression of the putative gene ppsγ, which is not located in the same gene cluster with ppsα and ppsβ, is lower in phenol-grown cells than in benzoate-grown cells. The enzyme activity of phenylphosphate synthase was observed in vitro in cell-free extracts of phenol-grown cells by measuring the conversion of phenol to phenylphosphate. Since the conversion of phenol to phenylphosphate can be accomplished without the presence of ppsγ, further verifications are needed to prove the participation of gene ppsγ in phenol degradation in D. anilini.
After activation of phenol to phenylphosphate, the latter is carboxylated to 4-hydroxybenzoate by phenylphosphate carboxylase. Homologues of three genes coding for the three subunits (ppcαβδ) of phenylphosphate carboxylase in T. aromatica are present in the genome of D. anilini. No homologue for ppcγ was found in the genome of D. anilini. The gene cluster under study carries only a homologue for the β-subunit of phenylphosphate carboxylase. The genes, ppcα, ppcβ, ppcδ, were all transcribed during growth on phenol. The expression of these genes except for ppcδ was observed in phenol-grown cells. However, attempts to demonstrate phenylphosphate carboxylase activity in cell-free extracts of phenol-grown cells failed. Earlier studies have shown, that the activity of phenylphosphate carboxylase in Thauera aromatica (earlier known as Pseudomonas strain K 172) is extremely oxygen sensitive and its activity in oxygen-saturated buffer had a half-life of 30 s [3, 7]. It is therefore likely, that phenylphosphate carboxylase is also present in D. anilini, but inactivated during cell-lysis despite all precautions made for excluding oxygen from the samples. The fact that all necessary enzymes for the degradation 4-hydroxybenzoate were expressed during growth with phenol leads to conclude that phenylphosphate, whose presence as an intermediate was proven, needs to undergo a carboxylation or carbonylation with phenylphosphate carboxylase as the most likely responsible enzyme.
4-hydroxybenzoate is subsequently transformed to the central intermediate benzoyl-CoA via 4-hydroxybenzoyl-CoA, the enzymes involved are 4-hydroxybenzoyl-CoA ligase and 4-hydroxybenzoyl-CoA reductase. The gene (locus tag H567DRAFT_02049) coding for 4-hydroxybenzoyl-CoA ligase in the genome of D. anilini shows homologies to genes coding for phenylphosphate-CoA ligase. The genes coding for 4-hydroxybenzoyl-CoA ligase and the α-subunit of 4-hydroxybenzoyl-CoA reductase are located in the gene cluster under study. This gene cluster carries a carbon monoxide dehydrogenase medium subunit (locus tag H567DRAFT_02055) and a carbon monoxide dehydrogenase small subunit (locus tag H567DRAFT_02054), which can catalyze the reduction of CO2 to CO. The resulting CO is combined with a methyl group to form acetyl-CoA by acetyl-CoA synthase through the Wood-Ljungdahl pathway in some anaerobic microbes [20, 21]. In D. anilini this enzyme system is needed for cleaving acetyl-CoA to CO and a methyl group to allow complete oxidation of the substrate to CO2 with sulfate as electron acceptor [22]. Since carbon monoxide dehydrogenase (CODH) was strongly expressed in phenol-grown cells in comparison to benzoate-grown cells (Fig. 4), CODH may also play a role in the upper phenol degradation pathway. For example, it could convert CO2 to CO and employ CO as a co-substrate to activate phenylphosphate, as reported in anaerobic acetone degradation by sulfate-reducing bacteria [23]. Another presumption is that CODH may have a reductive dehydroxylation function, by dehydroxylating 4-hydroxybenzoate or 4-hydroxybenzoyl-CoA to benzoate or benzoyl-CoA respectively. The function of 2, 3-dihydroxybenzoate-AMP ligase (locus tag H567DRAFT_02058), which is induced during growth with phenol, is unknown. In benzoate-grown cells, some of the genes in this gene cluster were still transcribed and translated. A possible reason may be that 10 generations of adaptation to benzoate are not enough to switch the metabolism from phenol to benzoate utilization completely.
The results obtained in this work support the hypothesis that sulfate-reducing bacteria use for phenol degradation a similar strategy as nitrate-reducing bacteria do. But it remains unclear how a sulfate reducer with its small energy budget can afford the high energy expenditure for phenol carboxylation and hydroxybenzoic acid activation which together require up to four ATP equivalents. The lack of ppcγ in the genome of D. anilini is in accordance with the results in the iron-reducing bacterium Geobacter metallireducens GS-15 [10], indicating a different catalytic mechanism of phenylphosphate carboxylase in sulfate-reducing and in iron-reducing bacteria. One possible activation mechanism that could be considered is a hypothetical phenylphosphate carbonylation to 4-hydroxybenzaldehyde with CO that is produced by the CO dehydrogenase located in the abovementioned gene cluster.
Conclusion
In this study, the genes coding for the enzymes involved in the anaerobic conversion of phenol to benzoyl-CoA were identified in the sulfate-reducing bacterium D. anilini. Analysis of the genome, transcriptome and proteome revealed that phenol is most likely activated to phenylphosphate, followed by carboxylation to 4-hydroxybenzoate, which is analogous to the well-known phenol degradation pathway in nitrate-reducing bacteria. Further evidence for phenylphosphate as an intermediate of phenol degradation in D. anilini is provided by in-vitro enzyme assays. Activity of phenylphosphate carboxylase could not be demonstrated by in-vitro enzyme assays, however all necessary genes for this enzyme complex were specifically expressed during growth with phenol. This work contributes to completing the picture of the phenol degradation pathway in anaerobic bacteria depending on different electron acceptors.
Methods
Bacterial strains and culture media
Desulfatiglans anilini DSM 4660 was isolated from marine sediments [24] and described as a sulfate-reducing bacterium oxidizing aniline. D. anilini was grown in bicarbonate-buffered (30 mM) and sulfide-reduced (2 mM) brackish water medium [25]. After autoclaving at 121 °C and 1 atm overpressure and cooling to room temperature under a stream of N2/CO2, 1 mL selenite tungstate solution/L and 1 mL 7 vitamins solution/L [26] were added. A heat-sterilized stock solution of a trace element mixture (SL 13) [27] was added to the basal medium (1:1000 v/v). Resazurin (0.4 mg mL− 1) was used as redox indicator. The pH was adjusted to 7.2. Benzoate or phenol was added anoxically from filter-sterilized anoxic stock solutions to 2 mM final concentration as growth substrate plus sulfate (10 mM) as terminal electron acceptor.
Adaptation of D. anilini to utilization of phenol or benzoate
D. anilini was inoculated into fresh medium with each 1 mM phenol or benzoate at an initial OD600 of around 0.04. The OD600 was monitored once per week until a maximum OD600 of around 0.18 was reached after 27 days. Cells were subsequently transferred to fresh medium four more times reaching over 135 days from initial culture.
Reverse transcription PCR analysis
For extracting RNA of D. anilini, 20 mL of phenol- or benzoate-grown cells in log-phase were collected by centrifuging at 11,700 x g for 20 min. Cells were washed with ultrapure water and centrifuged again. The pellets were used to extract RNA by using the TRIzol®Reagent with the PureLink®RNA Mini Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s protocol. RNA Clean & Concentrator Kit (ZYMO RESERACH, Irvine, California, USA) was used to remove contaminating DNA from RNA samples. The first strand cDNA was synthesized from total RNA using the SuperScript®III First-Strand Synthesis System for RT-PCR Kit (Invitrogen, Waltham, Massachusetts, USA) with random hexamer primers. Genomic DNA of D. anilini was isolated from 1.5 ml of a dense culture using the Gentra Puregene Cell Kit (Qiagen). The concentrations of DNA, RNA and cDNA were measured with a NanoDrop™ 2000c Spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
PCR amplification was performed using a T100 Thermal Cycler (Bio-Rad, Hercules, California, USA). The standard PCR mixture had a volume of 25 μL and contained 2.5 μL of 10 × PCR buffer, 5 nmol dNTPs, 50 pmol of each primer (Microsynth, Balgach, Switzerland), 4 nmol MgCl2, 0.2 μL Tag-polymerase (5 U/μL, Thermo Fisher Scientific, Waltham, Massachusetts, USA), and 2 μL cDNA or 10 to 50 ng genomic DNA as template. The PCR program consisted of an initial denaturation step at 94 °C for 3 min, followed by 31 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min, and a final elongation step of 72 °C for 5 min. Primer pairs used to amplify approximately 200–500 bp fragments of genes are listed in Table 2. The PCR purification products’ qualities were analyzed by electrophoresis in a 1.0% agarose gel at 110 V for 30 min.
Preparation of cell-free extracts
Cultures grown on phenol or benzoate were harvested at the end of the exponential growth phase (OD600 = 0.15–0.20) in an anoxic chamber (Coy, Ann Arbor, USA) by centrifugation (20,300 x g for 30 min at 4 °C, Dupont Sorvall, Midland, Canada). For total proteomics analysis, cells were washed twice by repeated centrifugation in anoxic 200 mL 50 mM potassium phosphate buffer containing 3 mM dithiothreitol, pH 7.5, and resuspended in 3–4 mL of the same buffer. Cells were broken anoxically by three passages through a cooled MiniCell French pressure cell (SLM Aminco, Cat. No. FA003, Urbana, Illinois, USA) operated at 137 MPa pressure. Cell debris was removed by centrifugation at 30,300 x g for 30 min at 4 °C to obtain the crude extract. The soluble protein fraction was obtained by ultra-centrifugation (150,000 x g for 60 min, Optima™ TL Ultracentrifuge, Beckman Coulter, Brea, California, USA) of the crude extract to remove insoluble membrane particles. The protein concentration was estimated with the Bradford assay using bovine serum albumin as protein standard [28].
Total proteomics analysis and database search
The supernatants containing soluble proteins were used for total proteomics analysis, whose concentrations were 2.7 mg mL− 1 (phenol-grown cells) and 2.9 mg mL− 1 (benzoate-grown cells), from which 500 μL supernatant was submitted to peptide-fingerprinting-mass spectrometry at the Proteomics Facility of the University of Konstanz. Total proteome analysis was performed using a LTQ Orbitrap Discovery with an Eksigent 2D-nano HPLC (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The mass spectrometry data was analyzed by the Mascot search engine [v2.2.2 from Matrix Science] [29], to identify and characterize proteins from the protein database of the IMG annotated genome of D. anilini. Quantitative analysis of the identified proteins was done by label-free quantification using the LFQ algorithms included in the Proteome Discoverer software package V1.3 (Thermo Scientific). Relative protein abundances were expressed as label-free quantification intensity (LFQ-intensity).
Phenylphosphate synthase activity
Cell extracts from cells grown on phenol or benzoate were prepared in 50 mM imidazole-HCl buffer (pH 7.0), 0.5 mM dithiothreitol, 0.5 mg DNase I, followed with a French press and ultra-centrifugation (30 min; 30,300 x g) to remove cell debris. The standard enzyme assay mixture (2 mL) for phenylphosphate synthase contained approximately 1 mg protein, 2 mM ATP, 2 mM MgCl2, 2 mM MnCl2, 2 mM KCl and 1 mM phenol. The enzyme tests were performed at 30 °C under strictly anaerobic conditions. To analyze the enzyme product, 300 μL samples were withdrawn at different time points and the reaction was stopped by addition of an equal volume of dichloromethane and centrifuged (11,700 x g for 10 min). The supernatant was transferred into 200 μL HPLC vials and analyzed by HPLC.
Phenylphosphate carboxylase activity
Cell extracts were prepared in 50 mM imidazole-HCl buffer (pH 7.0), 0.5 mM dithiothreitol, 10% glycerol, 0.5 mg DNase, using a French press, followed by ultra-centrifugation (30 min; 30,300 x g) to remove cell debris. The enzyme assays were performed at 30 °C under strictly anoxic conditions. The standard assay mixture (2 mL) contained 50 mM imidazole-HCl buffer (pH 7.0) with 0.5 mM dithiothreitol, 0.7 mg protein, 2 mM MgCl2, 2 mM MnCl2, 20 mM KCl, 1 mM phenylphosphate and 30 mM NaHCO3 (or 10% CO). The reaction products were monitored by UV/Vis absorption spectra or HPLC analysis. For spectrophotometric assays, 50 μL samples were withdrawn from the assay mixture at different time points, added into 950 μL of 1 M KOH solution and the absorption was determined at two wavelengths for quantification of phenylphosphate (phenolate ion at pH 14; 235 nm; ε = 9400 M− 1 cm− 1) and the product 4-hydroxybenzoate (280 nm; ε = 16,300 M− 1 cm− 1) [3]. For HPLC analysis, 300 μL samples were withdrawn from the assay mixture at different time points and the reaction was stopped by addition of an equal volume of acetonitrile and centrifuged (11,700 x g for 10 min). The supernatant (200 μL) was transferred to HPLC vials and analyzed by HPLC.
Analytical methods
The concentrations of phenol, phenylphosphate and 4-hydroxybenzoate were determined with a reversed-phase HPLC (Shimadzu, Kyoto, Japan) system equipped with a UV-visible diode array detector and a 4 um Max-RP 80 Å LC column (250*4.6 mm, Synergi) (Phenomenex, Torrance, California, USA) at 25 °C. Eluents were prepared by mixing ultrapure water with 0.1% H3PO4 (buffer B), and acetonitrile with 0.1% H3PO4 (buffer A) and filtration through 0.2 μm. A gradient of buffer B increasing from 80 to 60% was used at a flow rate of 1 mL min− 1. 50 uL samples were injected into the column. The compounds were identified by comparing the retention times and UV-spectra of peaks to the retention time and UV-spectra of the respective standards. Figure 5 was prepared by exporting the chromatogram data of the 200 nm PDA-channel from the Shimadzu LC solutions software to ASCII – format (time (s) and intensity (mAU)) and the time data was converted from s to min. Then, the data was converted to a Microsoft – Excel diagram to obtain a better resolution of the chromatogram layout.
Chemicals
All standard chemicals were of analytical quality and were obtained from Fluka (Buchs, Switzerland), Merck (Darmstadt, Germany) or Sigma (St. Louis, USA). Gases were purchased from Messer-Griesheim (Darmstadt, Germany) and Sauerstoffwerke Friedrichshafen (Friedrichshafen, Germany).
Abbreviations
- ASCII:
-
American Standard Code for Information Interchange
- AsrA:
-
dissimilatory adenylylsulfate reductase alpha subunit precursor
- BamA:
-
oxoenoyl-CoA hydrolase
- BamB-I:
-
benzoyl-CoA reductase
- BamQ:
-
hydroxyenoyl-CoA dehydrogenase
- BamR:
-
cyclohexadienoyl-CoA hydratase
- CODH:
-
carbon monoxide dehydrogenase
- D. anilini :
-
Desulfatiglans anilini DSM 4660
- G. metallireducens :
-
Geobacter metallireducens GS-15
- HPLC:
-
High-performance liquid chromatography
- IMG/M:
-
Integrated Microbial Genomes & Microbiomes
- LFQ:
-
Label-free quantification intensity
- MS:
-
mass spectrometry
- NCBI:
-
National Center for Biotechnology Information
- ORF:
-
Open reading frame
- ppc:
-
phenylphosphate carboxylase
- ppcα:
-
phenylphosphate carboxylase alpha subunit
- ppcβ:
-
phenylphosphate carboxylase beta subunit
- ppcγ:
-
phenylphosphate carboxylase gamma subunit
- ppcδ:
-
phenylphosphate carboxylase delta subunit
- ppsα:
-
phenylphosphate synthase alpha subunit
- ppsβ:
-
phenylphosphate synthase beta subunit
- ppsγ:
-
phenylphosphate synthase gamma subunit
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- T. aromatica :
-
Thauera aromatica; pps: phenylphosphate synthase
- UV/Vis:
-
Ultraviolet–visible spectroscopy
References
Kumar.A, Bisht.B.S, Joshi.V.D, Dhewa.T. Review on bioremediation of polluted environment: a management tool. Int J Environ Sci. 2011;1(6):1079–93.
Lack A, Fuchs G. Evidence that phenol phosphorylation to phenylphosphate is the first step in anaerobic phenol metabolism in a denitrifying Pseudomonas Sp. Arch Microbiol. 1994;161(2):132–9.
Lack A, Fuchs G. Carboxylation of phenylphosphate by phenol carboxylase, an enzyme-system of anaerobic phenol metabolism. J Bacteriol. 1992;174(11):3629–36.
Lack A, Tommasi I, Aresta M, Fuchs G. Catalytic properties of phenol carboxylase. In vitro study of CO2: 4-hydroxybenzoate isotope exchange reaction. Eur J Biochem. 1991;197(2):473–9.
Schmeling S, Narmandakh A, Schmitt O, Gad'on N, Schuhle K, Fuchs G. Phenylphosphate synthase: a new phosphotransferase catalyzing the first step in anaerobic phenol metabolism in Thauera aromatica. J Bacteriol. 2004;186(23):8044–57.
Narmandakh A, Gad'on N, Drepper F, Knapp B, Haehnel W, Fuchs G. Phosphorylation of phenol by phenylphosphate synthase: role of histidine phosphate in catalysis. J Bacteriol. 2006;188(22):7815–22.
Schühle K, Fuchs G. Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic phenol metabolism in Thauera aromatica. J Bacteriol. 2004;186(14):4556–67.
Biegert T, Altenschmidt U, Eckerskorn C, Fuchs G. Enzymes of anaerobic metabolism of phenolic compounds 4-hydroxybenzoate-CoA ligase from a denitrifying Pseudomonas species. Eur J Biochem. 1993;213(1):555–61.
Brackmann R, Fuchs G. Enzymes of anaerobic metabolism of phenolic compounds 4-hydroxybenzoyl-CoA reductase (dehydroxylating) from a denitrifying Pseudomonas species. Eur J Biochem. 1993;213(1):563–71.
Schleinitz KM, Schmeling S, Jehmlich N, von Bergen M, Harms H, Kleinsteuber S, Vogt C, Fuchs G. Phenol degradation in the strictly anaerobic iron-reducing bacterium Geobacter metallireducens GS-15. Appl Environ Microb. 2009;75(12):3912–9.
Zhang XM, Wiegel J. Reversible conversion of 4-hydroxybenzoate and phenol by Clostridium Hydroxybenzoicum. Appl Environ Microb. 1994;60(11):4182–5.
Gallert C, Winter J. Comparison of 4-hydroxybenzoate decarboxylase and phenol carboxylase activities in cell-free extracts of a defined, 4-hydroxybenzoate and phenol-degrading anaerobic consortium. Appl Microbiol Biot. 1992;37(1):119–24.
Ahn YB, Chae JC, Zylstra GJ, Haggblom MM. Degradation of phenol via phenylphosphate and carboxylation to 4-hydroxybenzoate by a newly isolated strain of the sulfate-reducing bacterium Desulfobacterium anilini. Appl Environ Microb. 2009;75(13):4248–53.
Breinig S, Schiltz E, Fuchs G. Genes involved in anaerobic metabolism of phenol in the bacterium Thauera aromatica. J Bacteriol. 2000;182(20):5849–63.
Heider J, Boll M, Breese K, Breinig S, Ebenau-Jehle C, Feil U, Gad'on N, Laempe D, Leuthner B, Mohamed ME, Schneider S, Burchardt G, Fuchs G. Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch Microbiol. 1998;170(2):120–31.
Wischgoll S, Heintz D, Peters F, Erxleben A, Sarnighausen E, Reski R, Van Dorsselaer A, Boll M. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol Microbiol. 2005;58(5):1238–52.
Buckel W, Kung JW, Boll M. The benzoyl-coenzyme a reductase and 2-hydroxyacyl-coenzyme a dehydratase radical enzyme family. Chembiochem. 2014;15(15):2188–94.
Peters F, Shinoda Y, McInerney MJ, Boll M. Cyclohexa-1,5-diene-1-carbonyl-coenzyme a (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J Bacteriol. 2007;189(3):1055–60.
Kuntze K, Shinoda Y, Moutakki H, McInerney MJ, Vogt C, Richnow HH, Boll M. 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme a hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ Microbiol. 2008;10(6):1547–56.
Wood HG. Life with CO or CO2 and H2 as a source of carbon and energy. FASEB J. 1991;5(2):156–63.
Ragsdale SW. Life with carbon monoxide. Crit Rev Biochem Mol. 2004;39(3):165–95.
Spormann AM, Thauer RK. Anaerobic acetate oxidation to CO2 by Desulfotomaculum acetoxidans. Demonstration of enzymes required for the operation of an oxidative Acetyl-CoA/ carbon monoxide dehydrogenase pathway Arch Microbiol. 1988;150(4):374–80.
Acosta OBG, Hardt N, Schink B. Carbonylation as a key reaction in anaerobic acetone activation by Desulfococcus biacutus. Appl Environ Microb. 2013;79(20):6228–35.
Schnell S, Bak F, Pfennig N. Anaerobic degradation of aniline and dihydroxybenzenes by newly isolated sulfate-reducing bacteria and description of Desulfobacterium-anilini. Arch Microbiol. 1989;152(6):556–63.
Schmidt A, Frensch M, Schleheck D, Schink B, Müller N. Degradation of acetaldehyde and its precursors by Pelobacter carbinolicus and P. acetylenicus. PLoS One. 2014;9(12)
Widdel F, Pfennig N. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty-acids .1. Isolation of new sulfate-reducing bacteria enriched with acetate from saline environments. Description of Desulfobacter Postgatei gen. Nov., Sp. Nov. Arch Microbiol. 1981;129(5):395–400.
Müller N, Griffin BM, Stingl U, Schink B. Dominant sugar utilizers in sediment of Lake Constance depend on syntrophic cooperation with methanogenic partner organisms. Environ Microbiol. 2008;10(6):1501–11.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67.
Acknowledgements
X. X. is grateful to China Scholarship Council (CSC) for providing a PhD scholarship. We thank Antje Wiese for preparing media, and Bernhard Schink for critically reading the manuscript. We are indebted to Andreas Marquardt, University of Konstanz, for the MS protein identification service. We also acknowledge the public service of the Integrated Microbial Genomes system (IMG) of the Joint Genome Institute (JGI) of the U.S. department of energy for making the genome sequence of Desulfatiglans anilini publicly available in the course of the one thousand microbial genomes (KMG-I) project. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Funding
The work of Xiaoman Xie was supported by the Chinese Scholarship Council.
Availability of data and materials
All data presented are included in the main article. The genome sequence of Desulfatiglans anilini DSM 4660 and its gene annotations are available on the Integrated Microbial Genomes database under the IMG genome ID 2526164742.
Author information
Authors and Affiliations
Contributions
XX and NM designed experiments, analyzed the data and wrote the manuscript. Experiments were conducted by XX. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. PCR products using genomic DNA as template. (PDF 73 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xie, X., Müller, N. Enzymes involved in the anaerobic degradation of phenol by the sulfate-reducing bacterium Desulfatiglans anilini. BMC Microbiol 18, 93 (2018). https://doi.org/10.1186/s12866-018-1238-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-018-1238-0