
Abstract
Expanding industrialization and the associated usage and production of mineral oil products has caused a worldwide spread of polycyclic aromatic hydrocarbons. These pollutants accumulate and persist under anoxic conditions but little is known about the biochemical reactions catalyzing their anaerobic degradation. Recently, carboxylation of naphthalene was demonstrated for the sulfate-reducing culture N47. Proteogenomic studies on N47 allowed the identification of a gene cluster with products suggested to be involved in the initial reaction of naphthalene degradation. Here, we performed comparative proteomic studies with N47 proteins extracted from naphthalene versus 2-methylnapththalene-grown cells on blue native PAGE. The analysis led to the identification of subunits of the naphthalene carboxylase of N47. Moreover, we show that the identified subunits are encoded in an operon structure within the previously mentioned naphthalene carboxylase gene cluster. These findings were supported by a pull-down experiment revealing in vitro interaction partners of a heterologously produced GST-tagged naphthalene carboxylase subunit. Based on these lines of evidence, naphthalene carboxylase is proposed to be a complex of about 750 kDa. Naphthalene carboxylase can be seen as a prototype of a new enzyme family of UbiD like de-/carboxylases catalyzing the anaerobic activation of non-substituted polycyclic aromatic hydrocarbons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous and persistent environmental pollutants originating from mineral oil products and spills, or from incomplete combustion. The high chemical stability and low aqueous solubility of PAHs make them very recalcitrant, especially in anoxic habitats (Meckenstock et al. 2016). Naphthalene represents the simplest polycyclic aromatic hydrocarbon and due to its relatively high solubility in water (240 µM at 25 °C) compared to larger molecular weight PAHs, it is often used as a model compound for studying PAHs degradation. In the last two decades, microbial degradation of naphthalene was reported under sulfate-reducing (Galushko et al. 1999; Meckenstock et al. 2000), iron-reducing (Kleemann and Meckenstock 2011; Lovley et al. 1994; Marozava et al. 2018), and methanogenic conditions (Christensen et al. 2004) (for review see Meckenstock and Mouttaki 2011).
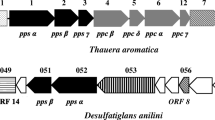
Metabolite studies with sulfate-reducing cultures revealed 2-naphthoic acid as a potential intermediate of anaerobic naphthalene degradation and the incorporation of [13C] bicarbonate into the carboxyl group indicated a direct carboxylation of naphthalene (Meckenstock et al. 2000; Zhang and Young 1997). More reduced metabolites such as tetrahydro-2-naphthoic acid suggested a subsequent ring reduction to initiate ring cleavage (Meckenstock et al. 2000). Later, proteogenomic studies on the sulfate-reducing cultures N47 and NaphS2 allowed the identification of gene clusters specifically induced by growth with naphthalene as electron and carbon source (Bergmann et al. 2011b; DiDonato et al. 2010). Some of the gene products of one cluster exhibited strong similarity to a gene encoding ubiquinone decarboxylase UbiD of Escherichia coli and subunits of phenylphosphate carboxylase, one of the key enzymes in anaerobic phenol degradation (Bergmann et al. 2011b; DiDonato et al. 2010; Schühle and Fuchs 2004). Furthermore, the putative naphthalene carboxylase genes showed homology to genes putatively coding for the anaerobic benzene carboxylase detected in the iron-reducing enrichment culture BF (Abu Laban et al. 2010) (Fig. 1). Recently, Mouttaki et al. (2012) provided the first biochemical evidence confirming the naphthalene carboxylation reaction in the sulfate-reducing culture N47.

Organization of gene clusters encoding UbiD-like proteins involved in anaerobic degradation of aromatic compounds. Open reading frames are represented by arrows. UbiD-like genes are shown in a pattern fill, whereas striped arrows represent PpcA-like genes. PpcD-like genes are indicated by dark grey and UbiX-like genes in light grey. Further details are mentioned in the text
In order to identify the subunits of the naphthalene carboxylase protein complex, a recombinant production of the putative subunits was attempted in various E. coli strains with differing protein tags but yielded, for the most part, insoluble protein. During attempts to purify the naphthalene carboxylase complex under anaerobic conditions directly from N47 cells, all enzyme activity was lost (Kölschbach, unpublished results).
As such, it was unclear which of the gene products found to be differentially abundant in the naphthalene grown N47 culture, were involved in the naphthalene carboxylase protein complex (Bergmann et al. 2011b).
The aim of the present study was, thus, to identify which genes belong to the putative naphthalene carboxylase operon and which polypeptides constitute the naphthalene carboxylase protein complex of N47.
Materials and methods
Growth of culture N47 and preparation of cell-free extracts
The sulfate-reducing culture N47 was cultivated at 30 °C in serum bottles containing bicarbonate-buffered freshwater medium using naphthalene as carbon and electron source and sulfate as electron acceptor (Meckenstock et al. 2000). Naphthalene or 2-methylnaphthalene was added as a 1.5% (w/v) solution in 2,2,4,4,6,8,8-heptamethylnonane (20 mL/L culture volume). For the preparation of cell-free extracts, cells were harvested by centrifugation (3200×g, 4 °C, 30 min) under anoxic conditions in an anaerobic chamber (Labstar, MBRAUN, Garching, Germany) under 100% N2 atmosphere. The anaerobic chamber, where all anaerobic experiments took place, is equipped with an internal O2 sensor to ensure that the oxygen concentration does not reach levels above 3 ppm. The cells were harvested in the mid-exponential growth phase, which had been determined in previous experiments (Meckenstock et al. 2000; Musat et al. 2009), after 6 weeks of cultivation. The cells were disrupted by 3–4 cycles of sonication (15–20 s/cycle; 30 kHz; 50% pulse; 40% amplitude; UP50H, Hielscher Ultrasonics GmbH, Teltow, Germany) in a pre-chilled metallic block followed by a 20 min centrifugation step (13,000×g, 4 °C). The supernatant was amended with 1 × complete protease inhibitor cocktail (Roche, Basel, Switzerland) to prevent protein degradation.
Operon mapping (transcriptional analysis)
RNA extraction
N47 cells were harvested by centrifugation of 150 mL culture. The cell pellets were resuspended in a buffer containing 50 mM Na-Acetate, pH 5.3 and 10 mM Na-EDTA. RNA was extracted by bead beating in the presence of sodium phosphate buffer and sodium dodecyl sulfate as reported by Schmitt et al. (1990). After centrifugation (5 min, 13,000×g, 4 °C), the supernatant was extracted with equal volumes of phenol-chloroform-isoamylalcohol [24:24:1 (v/v/v)] and chloroform-isoamylalcohol [24:1 (v/v)] prior to precipitation (30 min, 13,000×g, 4 °C) with 2 µL glycogen (Roche, Basel, Switzerland) and 2 volumes of polyethylene glycol (Griffiths et al. 2000). Subsequently, the nucleic acid pellet was washed with 70% ethanol and resuspended in RNase-free water (Promega, Fitchburg, WI). The co-extracted DNA was digested with RQ1 RNase-free DNase (Promega, Fitchburg, WI) according to the manufacturer’s instructions. The RNA was visualized by standard agarose gel electrophoresis and analyzed using NanoDrop (NanoDrop Technologies, Inc., Wilmington, DE) to estimate the quantity. RNA was stored at − 80 °C until further use.
RT-PCR
The RNA was reverse transcribed to cDNA by GoScript™ reverse transcriptase using random hexamer primers (0.5 µg/reaction; Thermo Fisher Scientific, Waltham, MA) or gene-specific reverse primers (Table 1; 50199r, 48240r, 46360r, 44481r; 5 pmol each/reaction) as described by the manufacturer’s protocol (Promega, Fitchburg, WI). These primers were designed manually and synthesized by Eurofins Genomics (Ebersberg, Germany). As negative controls, reactions were performed without the reverse transcriptase to detect eventual residual genomic DNA contamination.
PCR amplification
Intergenic regions were amplified via PCR using the primer sets listed in Table 1 with the following program: 5 min at 94 °C initial denaturation, 35 cycles of amplification (30 s at 94 °C, 15 s at 55 °C, 1 kb/60 s at 72 °C) and 5 min at 72 °C of final extension. The PCR reaction used 1 × Taq PCR buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.5 μM primer (Eurofins Genomics, Ebersberg, Germany) and 1.25 U Taq polymerase (Thermo Fisher Scientific, Waltham, MA).
Cloning, production and purification of GST-NcA
Genomic DNA from the sulfate-reducing culture N47 was extracted with the QIAamp DNA Mini kit (Qiagen, Hilden, Germany) as described by the manufacturer. Gene N47_K27540 encoding one of the naphthalene carboxylase subunits (NcA) (Bergmann et al. 2011b), was amplified from N47 genomic DNA using the primer set ncA_for (GTAGTCGACTGATGGCGTTTAAAGATTTGAG) and ncA_rev (GTAGCGGCCGCGAATCCATATTTATTCCAATTG). The PCR product was purified using the PCRextract kit (5Prime, Hamburg, Germany) according to the manufacturer’s protocol. Standard molecular cloning procedures were applied. Enzymes were purchased from Thermo Fisher Scientific. The amplicon was inserted into the expression vector pGEX-6P-1 (GE Healthcare Europe, Freiburg, Germany) containing an IPTG-inducible lac-promotor and a GST-tag. Chemically competent E. coli Rosetta™ 2(DE3)pLysS (Merck, Darmstadt, Germany) were transformed with the plasmid pGEX-6P-1_ncA as described previously (Inoue et al. 1990). For production of the GST-tagged fusion protein GST-NcA, 1 L of LB-medium (Carl Roth®, Karlsruhe, Germany) containing ampicillin (100 µg/mL) was inoculated with the recombinant E. coli Rosetta™ 2(DE3)pLysS including the plasmid pGEX-6P-1_ncA and incubated at 37 °C. Gene expression was induced by addition of 0.5 mM IPTG when the culture reached an optical density of 0.5 at 578 nm, and the cells were shifted to room temperature for an additional 3 h. The cells were harvested at 3200×g for 15 min at 4 °C and the pellet was stored at − 20 °C until further use. GST-NcA was purified using batch purification with Glutathione Sepharose 4B beads as recommended by the manufacturer with the following modifications (GE Healthcare Europe, Freiburg, Germany). The stored pellet was resuspended in binding buffer (50 mM Tris/HCl, pH 7.5; 1 M NaCl; 1 mM EDTA) and the cells were disrupted by sonication as described above. After sonication, 1% IGEPAL® CA-630 (Sigma-Aldrich, St. Louis, MO) was added. For purification, 1 mL of Glutathione Sepharose 4B slurry was used. The beads were incubated with E. coli Rosetta™ 2(DE3)pLysS cell-free extract containing GST-NcA on an end-over-end rotator at 4 °C for 2 h and subsequently 3 times washed with washing buffer (50 mM Tris/HCl, pH 7.5; 140 mM NaCl). The GST-NcA coupled to the Sepharose beads was used for subsequent pull-down experiments. The purified protein was stored in 20 mM Tris/HCl, pH 7.5 with 50% glycerol at − 20 °C.
In vitro protein interaction analysis (pull-down experiment)
Purified GST-NcA coupled to the Glutathione Sepharose beads was washed with 1x PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3) and 120 µL of the slurry was incubated for 4 h at 4 °C with 500 µL of the respective cell-free extract. After three washing steps (1 × PBS, 5 min, 4 °C), 30 µL beads were combined with 5 × SDS-PAGE loading dye and analyzed via SDS-PAGE. Protein bands were excised from the SDS-PAGE and submitted for proteomic analysis.
Blue native PAGE
Blue native PAGEs (BNPs) were performed in an anoxic chamber with N2-atmosphere using NativePAGE™ Novex® 4–16% Bis–Tris gels in the XCell SureLock® Mini Cell (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. Activity of the naphthalene carboxylase was confirmed by an activity assay before samples were loaded on the gel. The activity assay contained a saturated naphthalene solution, 25 mM NaHCO3 and 5 mM ATP in 100 mM MOPS/KOH, 15 mM MgCl2, pH 7.3 buffer. The assay was started by the addition of cell-free extract and stopped after 0, 15, 45, 90 min with 10% formic acid.
Proteomic analysis
In-gel tryptic digest
Coomassie-stained excised gel-pieces were digested as described previously (Merl et al. 2012). The eluted peptides were dried in a speed vac and stored at − 20 °C until further use.
Mass spectrometric measurements
Dried digested samples were thawed and dissolved in 45 µL of 2% acetonitrile/0.5% trifluoroacetic acid. After brief centrifugation (13,000×g, 15 min), LC–MS/MS analysis was performed on an Ultimate3000 nano HPLC system (Dionex, Sunnyvale, CA) online coupled to a LTQ OrbitrapXL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) by a nano spray ion source as described previously (Hauck et al. 2010; Merl et al. 2012). The samples were analyzed with and without the addition of dimethylsulfoxide (DMSO) to the buffers. Samples were loaded onto the C18 trap column at a flow rate of 30 μL/min in 7% acetonitrile/0.1% formic acid (3% buffer B (73% ACN/3% DMSO/0.1% formic acid (FA) in HPLC‐grade water) and 97% buffer A (2% ACN/3% DMSO/0.1% FA)) (Hahne et al. 2013). After 5 min, the peptides were eluted from the trap column and separated on the analytical column by a 135 min gradient from 7 to 32% acetonitrile in 0.1% formic acid (3 to 35% of buffer B) at 300 nL/min flow rate followed by a short gradient from 32 to 93% acetonitrile in 0.1% formic acid (35 to 95% buffer B) in 5 min. Between each sample, the gradient was set back to 7% acetonitrile in 0.1% formic acid (3% buffer B) and left to equilibrate for 20 min.
From the MS pre-scan, the 10 most abundant peptide ions were selected for fragmentation in the linear ion trap if they exceeded an intensity of at least 200 counts and if they were at least doubly charged. During fragment analysis a high-resolution (60,000 full-width half maximum) MS spectrum was acquired in the Orbitrap with a mass range from 300 to 1500 Da.
Label-free quantitative analysis
The acquired raw data of each dataset was loaded in the Progenesis LC–MS software (version 2.5, Nonlinear) for label free quantification and analyzed as described previously (Hauck et al. 2010; Merl et al. 2012). Briefly, profile data of the MS scans were transformed to peak lists with respective peak m/z values, intensities, abundances (areas under the peaks) and m/z width. MS/MS spectra were treated similarly. After reference selection, the retention times of the other samples were automatically aligned to a maximal overlay of all features. Features with only one charge or more than seven charges were excluded from further analyses. After normalization and assignment of the samples to the respective groups, all MS/MS spectra were exported as Mascot generic file (mgf) and used for peptide identification with Mascot (version 2.4) in the N47 protein database (version 4, 1302846 residues, 5001 sequences). Search parameters used were: 10 ppm peptide mass tolerance and 0.6 Da fragment mass tolerance, one missed cleavage allowed, carbamidomethylation was set as fixed modification, methionine oxidation and asparagine or glutamine deamidation were allowed as variable modifications. Searches were performed with a Mascot ion score cut-off of 30 and an appropriate significance threshold p, in order to reach a maximum false discovery rate of 1%. Search results were re-imported into the Progenesis LC–MS software. The abundances of all peptides allocated to each protein were summed up and exported. The resulting normalized protein abundances were used for calculation of fold-changes of proteins and for calculation of significance using an unpaired both-sided Student’s t test in Excel.
Protein determination
Protein concentrations were determined by the method of Bradford (1976) using Quick Start™ Bradford 1 × Dye Reagent (Bio-Rad Laboratories, Hercules, CA) and bovine serum albumin as standard.
SDS-PAGE
SDS polyacrylamide gel electrophoresis was performed as described previously (gel concentration 15%) (Laemmli 1970). PageRuler prestained protein ladder was used as a marker (Thermo Fisher Scientific, Waltham, MA).
Results
Determining the structure of the naphthalene carboxylase operon
Messenger RNA was extracted from N47 cells grown with naphthalene in order to identify genes of the putative naphthalene carboxylase gene cluster belonging to a transcriptional unit. The RNA was reverse transcribed into cDNA and PCR was performed using primers covering genes and/or intergenic regions to identify the potential operon (Table 1). Two genes were assigned to belong to the same operon (same mRNA) if a forward and a reverse PCR primer hybridizing to one of two adjacent genes, respectively, produced a PCR amplicon. Gene annotations were taken from Bergmann et al. (2011b). Amplicons were obtained between genes N47_K27540 and N47_K27520, N47_K27520 and N47_K27500, N47_K27500 and N47_K27480, and between N47_K27480 and N47_K27460. The PCR-products indicated that the naphthalene carboxylase gene cluster of strain N47 is transcribed as an operon of around 8.9 kb containing 9 genes from genes N47_K27540 to N47_K27460 (Fig. 2).

a Organization of the gene cluster encoding enzymes potentially involved in the initial carboxylation reaction of naphthalene degradation in the sulfate-reducing Deltaproteobacteria N47 (GI:308273914 to GI:308273898). Open reading frames which products show similarity to UbiD-like carboxylases are filled in black, non-carboxylase-like subunits are shown in grey. Genes which are co-transcribed are indicated by a pattern fill. ORFs which products share high homology between the two strains have the same filling. Arrows represent primers used for transcriptional analysis by gene-specific PCR. Annotated function of the gene products: 1, putative phenylphosphate carboxylase, alpha subunit; 2, putative phenylphosphate carboxylase, gamma subunit; 3, MRP, Fer4_NifH superfamily; 4, ParA/MinD ATPase like, MRP, Fer4_NifH superfamily; 5, UbiD family decarboxylase; 6, conserved hypothetical protein; 7, UbiD family decarboxylase; 8, conserved hypothetical protein; 9, UbiD family decarboxylase; 10 & 11, HAD hydrolase; 12, membrane protein involved in aromatic hydrocarbon degradation; 13, IS4 transposase; 14, succinate dehydrogenase and fumarate reductase iron-sulfur protein; 15, putative succinate dehydrogenase flavoprotein subunit; 16, UbiD family decarboxylase; 17, pyridoxamine 5′-phosphate oxidase family protein. b Transcriptional analysis by RT- and subsequent gene-specific PCR. Results of gene-specific PCRs using three different templates per primer set. A 1.5% agarose gel stained with GelRed was used. M, DNA ladder; numbers, name of used primer set; lane 1, gene-specific PCR on cDNA template (addition of reverse transcriptase during cDNA synthesis); lane 2, control for potential genomic DNA contamination in cDNA template (no addition of reverse transcriptase during RT-PCR); lane 3, gene-specific PCR on genomic DNA (positive control). Random hexamer primers were used in the RT-PCR
Determining the structure of the naphthalene carboxylase protein complex
Blue native PAGEs (BNPs) and LC–MS/MS analysis of the bands were performed to determine the size and polypeptide composition of the naphthalene carboxylase complex. Bands were excised from the gel and digested in-gel with trypsin. Then, the samples were dried and LC–MS/MS analysis was performed to determine the peptide composition of the different bands. The BNPs were performed under anoxic conditions to ensure that molecular oxygen did not impair the native conformation of the protein complexes.
Cell-free extracts of N47 cells grown with naphthalene exhibited a prominent band with a size of 750 kDa (Fig. 3), which was not present in cells grown with 2-methylnaphthalene. Furthermore, two prominent bands of around 560 kDa and 400 kDa were more intense from cells grown with naphthalene (Fig. 3, Suppl. Table 1).

Differential protein induction analysis of N47 on blue native PAGE. Boxes indicate slices of the gel which were cut out for proteomic analysis. Lane 1, cell-free extract of naphthalene-grown N47 cells; lane 2, molecular mass standard (1236–66 kDa); lane 3, cell-free extract of 2-methylnaphthalene-grown N47 cells
The three regions of the gels showing the differentially induced bands with sizes of about 750, 560, and 400 kDa were excised and analyzed by label-free quantitative proteomics (Suppl. Table 2). The 750 kDa band contained a total of 288 proteins, 29 of which were differentially abundant with a ratio of ≥ 2 in cells grown with naphthalene compared to 2-methylnaphthalene (Suppl. Table 2A). The ratio was defined as the protein ratio calculated by normalized abundances from naphthalene to 2-methylnaphthalene samples. Furthermore, for identification a p-value of ≤ 0.01 and a confidence score of > 30 were applied as cut-off. In all three bands excised from the gel, the most abundant polypeptides were UbiD carboxylase-like gene products of the putative carboxylase gene cluster (Suppl. Table 2A–C). Almost all of the gene products encoded in the naphthalene carboxylase operon were detected in the 750 kDa protein complex. Only the gene product N47_K27460 was not found. This gene product showed high similarity to PpcX, a phenol-induced UbiD like protein from T. aromatica and A. aromaticum, which is encoded in the phenol gene cluster but is not part of the phenylphosphate carboxylase complex (Schühle and Fuchs 2004). Three UbiD-like proteins N47_K27540 (52.4 kDa), N47_K27500 (57 kDa) and N47_K27480 (54.5 kDa) were part of the naphthalene carboxylase complex. Furthermore, N47_K27530 (9.8 kDa) which contained no conserved domains and the ParA/MinD ATPase-like proteins N47_K27510 (29.4 kDa) and N47_K27520 (33.6 kDa) were more abundant in naphthalene-grown N47 cells and therefore are proposed to represent subunits of the complex. Moreover, N47_K27490 (9.2 kDa) as well as N47_K27470 (16.6 kDa) were identified and both annotated as hypothetical proteins.
In the 560 kDa band, 23 proteins among 133 detected were differentially abundant with naphthalene as substrate (Suppl. Table 2B). The most abundant and differentially induced proteins were the carboxylase-like subunits N47_K27540, N47_K27500, N47_K27480 and N47_K27530 together with the heterodisulfide reductase subunits (N47_J04330, N47_J04340, N47J04350 and N47_G39330, N47_G39340). Moreover, N47_E46900 a protein annotated as K(+)-insensitive pyrophosphate-energized proton pump was detected and differentially produced.
In the 400 kDa band, 10 polypeptides out of 125 detected were differentially abundant in naphthalene-grown cells (Suppl. Table 2C). The most abundant and at the same time differentially produced proteins were represented by the carboxylase-like proteins N47_K27500 and N47_K27480. The ATPase-like protein N47_K27520 as well as proteins annotated as heterodisulfide reductase subunits (N47_J04330, N47_J04340, N47J04350) were differentially abundant. The K(+)-insensitive pyrophosphate-energized proton pump N47_E46900 was again identified.
In vitro protein interaction assays
In vitro interaction pull down assays were performed in order to identify subunits of naphthalene carboxylase interacting with the phenyl phosphate carboxylase (ppc)-like polypeptide N47_K27540 (Fig. 4, Suppl. Fig. 1), encoded by the first gene of the naphthalene carboxylase operon (Fig. 2a).

SDS-PAGE of a protein interaction assay with GST-NcA in cell-free extracts of N47 grown with naphthalene and 2-methylnaphthalene. Lane 1, purified GST-NcA coupled to glutathione Sepharose beads (input sample, 30 µL); lane 2, purified GST-NcA coupled to glutathione Sepharose after 4 h of incubation with cell-free extract of naphthalene-grown N47 cells (30 µL); lane 3, purified GST-NcA coupled to glutathione Sepharose beads (lane 1) after 4 h of incubation with cell-free extract of 2-methylnaphthalene-grown N47 cells (30 µL); lane 4, molecular mass standard (170, 130, 100, 70, 55, 40, 35, 25, 15 and 10 kDa); lane 5, GST coupled to glutathione Sepharose beads after 4 h of incubation in cell-free extracts of naphthalene-grown N47 cells; lane 6, GST coupled to glutathione Sepharose beads after 4 h of incubation in cell-free extracts of 2-methylnaphthalene-grown N47 cells. Vertical boxes indicate parts of the gel which were cut and send for proteomic analysis
The most abundant proteins interacting with GST-NcA from naphthalene compared to 2-methylnaphthalene-grown cells were the carboxylase-like proteins N47_K27500 and N47_K27480 with enrichment factors of 23.6 and 31.0, respectively (Table 2A, B), and the two ATPase-like proteins N47_K27520 and N47_K27510 with enrichment factors of 50.4 and 56.2 (Table 2D). These proteins were also identified in the native conformation of the naphthalene carboxylase complex analyzed with blue native gels. Furthermore, the carboxylase-like protein N47_K27460 showed interaction with GST-NcA. The corresponding gene is also part of the naphthalene carboxylase operon but the polypeptide could never be detected in the blue native gels. Moreover, N47_K27400 a succinate dehydrogenase-like flavoprotein was identified in one of the sliced bands with an enrichment factor of 44.6 (Suppl. Table 1).
The genes of several other proteins which are not part of the naphthalene carboxylase gene cluster also interacted in the pull-down assay (Table 2). For example, N47_A07150, a FAD-binding domain protein, was enriched with a factor of 103.2 (Table 2C). Moreover, the ATPase-like protein N47_K27600, which was also detected in the 750 kDa band of the blue native gels, interacted with an enrichment factor of up to 84.9 (Table 2C, D). Furthermore, a sulfate adenylyltransferase (N47_J04360) showed an enrichment factor of 30.6 (Table 2D). This protein was not identified in the blue native gels but it is encoded in close vicinity to the putative heterodisulfide reductase subunits N47_J04330, N47_J04340 and N47_J04350, which showed up in naphthalene grown cells in the blue native gels.
Discussion
The carboxylation of naphthalene is an unprecedented biochemical reaction. In proteogenomic and transcriptomic analyses a conserved gene cluster encoding carboxylase-like proteins was identified in the sulfate-reducing culture N47 (Bergmann et al. 2011b; DiDonato et al. 2010). Due to differential abundance with naphthalene, the gene products were proposed to be involved in the carboxylation of naphthalene. In the present study, we analyzed the structure of the respective operon and the composition of the naphthalene carboxylase protein complex.
Organization of the naphthalene carboxylase operon
The transcriptional analysis of the putative naphthalene carboxylase gene cluster revealed that the naphthalene carboxylase is transcribed as one operon ranging over 9 genes from N47_K27540 to N47_K27460. It contains the genes N47_K27540, N47_K27500, N47_K27480 and N47_K27460 encoding putative UbiD carboxylase like subunits. In E. coli, UbiD catalyzes the decarboxylation of an isoprenylated 4-hydroxybenzoate derivative in ubiquinone biosynthesis (Cox et al. 1969; Meganathan 2001). The identified subunits in N47 showed up to 46% sequence identity to the alpha subunit of the phenylphosphate carboxylase PpcA of T. aromatica and up to 48% to a putative anaerobic benzene carboxylase AbcA of the iron-reducing enrichment culture BF (Abu Laban et al. 2010; Schühle and Fuchs 2004). Two ParA/MinD ATPase-like proteins, N47_K27510 and N47_K27520, are encoded in the putative naphthalene carboxylase operon. Proteins with a ParA/MinD domain are involved in plasmid segregation and cell division (Gerdes et al. 2000) but are also found in iron-sulfur cluster biosynthesis (ApbC/Nbp35 homologs). More recent studies showed that the ParA/MinD family is further involved in the positioning of large cytoplasmic protein complexes such as carboxysomes or chemotaxis clusters (Lutkenhaus 2012). A similar function of the proteins N47_K27510 and N47_K27520 is possible for the quite large carboxylase complex. Additionally, the genes N47_K27530, N47_K27490, and N47_K27470 annotated as hypothetical proteins are encoded within the naphthalene carboxylase operon. Due to the small size and the lack of conserved domains these genes might encode linker proteins of the naphthalene carboxylase complex.
Two subsequent genes were not part of the operon, N47_K27450 and N47_K27440. They are similar to genes coding for HAD-like polypeptides (cd01427; conserved domain database, National Center for Biotechnology Information) (Bergmann et al. 2011a; Selesi et al. 2010). The HAD-like superfamily is composed of hydrolases comprising phosphatases, among others. For example, the HAD-like protein PpcC is part of the phenylphosphate carboxylase in Thauera aromatica, a key enzyme in anaerobic phenol degradation (Schühle and Fuchs 2004). PpcC was proposed to bind and dephosphorylate the substrate phenylphosphate. As there was no indication for such a phosphorylated intermediate in the carboxylation of naphthalene, a phosphatase-like subunit might not be required in the functional naphthalene carboxylase complex.
Furthermore, the naphthalene carboxylase gene cluster was surrounded by viral genes in N47 (N47_K27300, N47_K27370, N47_K27640 putative transposases). Considering the high sequence similarity of the gene clusters and the presence of genetic elements which are involved in gene mobility, horizontal gene transfer of the genes for naphthalene degradation is likely.
Naphthalene carboxylase complex and its subunits
In comparative native proteomic studies with blue native gels, three prominent naphthalene-induced bands with a size of around 750, 560, and 400 kDa were identified. Almost all polypeptides detected in the 750 kDa band, which were differentially abundant with naphthalene versus 2-methylnaphthalene as growth substrate, were encoded in the naphthalene carboxylase operon. The prominent bands at 560 and 400 kDa contained less polypeptides indicating disassociated sub complexes. This is supported by the fact that the 750 kDa protein complex was only detectable from freshly harvested cells grown with naphthalene as carbon and electron source. The 750 kDa band was absent in 2-methylnaphthalene-grown cells where naphthalene carboxylase activity could hardly be detected. Thus, our indirect evidence indicates that the 750 kDa band most likely represents the native naphthalene carboxylase complex. At the present state however, we were not able to purify an active naphthalene carboxylase enzyme complex. Considering the proteins contained in the 750 kDa band, the native naphthalene complex putatively consists of three UbiD-like carboxylase subunits N47_K27540 (52.4 kDa), N47_K27500 (57 kDa) and N47_K27480 (54.4 kDa), the ParA/MinD ATPase-like proteins N47_K27510 (29.4 kDa) and N47_K27520 (33.6 kDa). Furthermore, the subunits N47_K27530 (9.8 kDa), N47_K27490 (9.2 kDa), and N47_K27470 (16.6 kDa) are part of the naphthalene carboxylase complex as putative linker proteins.
Therefore, all subunits encoded in the naphthalene carboxylase operon, except for the carboxylase-like subunit N47_K27460, seem to be part of the naphthalene carboxylase complex. Our blue native gel experiments indicated that the naphthalene carboxylase complex has a size of around 750 kDa in native conformation. If all the subunits encoded in the naphthalene carboxylase operon would be involved in the complex only once, it would have a size of around 314 kDa. Without the UbiD-like subunit N47_K27460, which might not be part of the naphthalene carboxylase, the complex is around 263 kDa.
Similar to the naphthalene carboxylase complex, the phenylphosphate carboxylase from T. aromatica represents a new type of UbiD-like carboxylase (Schühle and Fuchs 2004). The purified enzyme is composed of four subunits- of two UbiD-like proteins (αβ, 54 and 53 kDa), a hypothetical protein (γ, 10 kDa) and a phosphatase-like protein (δ, 18 kDa). The authors suggested a trimeric structure (αβγδ)3 for this carboxylase complex.
Assuming a similar trimeric structure analogous to the phenylphosphate carboxylase, the naphthalene carboxylase complex would have a molecular mass around 943 kDa or 787 kDa with or without N47_K27460, respectively. The latter matches the observed molecular mass of 750 kDa.
Interaction of naphthalene carboxylase with other proteins
The carboxylase-like protein N47_K27460 was encoded within the naphthalene carboxylase operon and an interaction with the other carboxylase-like proteins was shown in a pull-down assay, but it was not detected within the naphthalene-induced bands on BNPs. The interaction between the carboxylase complex and N47_K27460 could have either been destroyed during sample preparation or N47_K27460 is not part of the naphthalene carboxylase complex. The UbiD-like protein N47_K27460 was also not detected on 2D PAGEs (Bergmann et al. 2011b). In a phylogenetic tree of UbiD/PpcA-like subunits involved in anaerobic degradation of aromatic compounds, the protein formed a cluster with PpcX from T. aromatica and A. aromaticum strain EbN1 (Suppl. Fig/ 2, Suppl. Table 3). PpcX is a phenol-induced UbiD-like protein and its gene is encoded within the phenol gene cluster but it is not part of the phenylphosphate carboxylase complex (Schühle and Fuchs 2004).
Moreover, proteins which are annotated as heterodisulfide reductases were identified within the naphthalene-induced bands in BNPs. Heterodisulfide reductase-like proteins are also involved in anaerobic degradation of monoaromatic compounds. The BamDE subunits of the class II benzoyl-CoA reductase of Geobacter metallireducens share sequence similarity to heterodisulfide reductases (Fuchs et al. 2011; Kung et al. 2009). Here, benzoyl-CoA is reduced in an electron bifurcation process via ferredoxinred coupled to the exergonic reduction of NAD(P)+ using another molecule of the same ferredoxinred. However, involvement of a reduction step to carboxylate naphthalene is not implicit and the appearance of heterodisulfide reductase-like proteins cannot be explained at the present state of knowledge.
UbiD-like (de)carboxylases involved in anaerobic degradation of aromatic compounds
In E. coli, the enzymes UbiD and UbiX facilitate the decarboxylation of 3-octaprenyl-4-hydroxybenzoate to 2-octaprenylphenol in ubiquinone biosynthesis (Gulmezian et al. 2007; Zhang and Javor 2003). Recently, UbiX and UbiD were identified as flavin mononucleotide (FMN) containing proteins working in a two-component system. UbiX synthesizes a prenylated FMN by adding a non-aromatic fourth ring to the flavin mononucleotide. UbiX enzyme activity is independent from metal cofactors and utilizes dimethylallyl-monophosphate as prenylating substrate (Payne KAP 2015; White et al. 2015). The prenylated FMNpr is transferred to UbiD, which performs the decarboxylation reaction.
In the N47 genome, a UbiX-like gene was not part of the naphthalene carboxylase cluster but two completely identical copies N47_B20630 and N47_H21380 were encoded elsewhere in the genome. In proteomic analyses, differentiation between these two proteins was not possible but one or both were differentially induced in cells grown with naphthalene.
At the present state of knowledge, the presence of a cofactor in the naphthalene carboxylase remains elusive. However, the amino acid sequences suggested potential binding sites for FMN in the carboxylase-like subunits N47_K27540, N47_K27500, N47_K27480, N47_K27460 (GO0010181; UniProt-GO annotation database, European Bioinformatics Institute) and nucleotide-binding domains in the ATPase-like proteins N47_K27510 and N47_K27520 (cd02037; conserved domain database, National Center for Biotechnology Information).
Various UbiD-like (de)carboxylases involved in the degradation of aromatic compounds have been identified in recent years (Fig. 1). A gene cluster putatively encoding enzymes involved in the degradation of terephthalate was identified. One gene product showed similarity to an UbiD-like protein and was suggested to be a terephthalate decarboxylase (Lykidis et al. 2011; Wu et al. 2013). Therefore, analogous to the synthrophic, terephthalate-degrading Pelotomaculum spp. the authors proposed that terephthalate is first activated by a ligase and subsequently decarboxylated by a complex of an UbiD-like protein and a phenylphosphate γ-subunit-like protein to produce benzoyl-CoA. The UbiD-like subunit showed more than 40% sequence identity to N47_K27540. This reaction could recently be shown with phthalate as substrate in cell extracts of the strains Thauera, Azoarcus and ‘Aromatoleum’ (Ebenau-Jehle et al. 2017; Junghare et al. 2016).
Conclusion
Non-substituted PAHs like naphthalene need an initial activation in order to be further degraded. This first important reaction is carried out by a carboxylase in the case of naphthalene (Mouttaki et al. 2012). The naphthalene carboxylase complex consists of eight proteins, which are encoded in one operon. The protein N47_K27460 is also encoded in the operon but is not part of the complex. Comparison to the phenylphosphate decarboxylase suggests a similar trimeric structure for the naphthalene carboxylase complex with a size of 750 kDa. Three of the subunits building the protein complex were identified as UbiD-like proteins, putting the naphthalene carboxylase in the growing family of de-/carboxylases using a prenylated flavin cofactor.
References
Abu Laban N, Selesi D, Rattei T, Tischler P, Meckenstock RU (2010) Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ Microbiol 12:2783–2796
Bergmann F, Selesi D, Weinmaier T, Tischler P, Rattei T, Meckenstock RU (2011a) Genomic insights into the metabolic potential of the polycyclic aromatic hydrocarbon degrading sulfate-reducing Deltaproteobacterium N47. Environ Microbiol 13:1125–1137
Bergmann FD, Selesi D, Meckenstock RU (2011b) Identification of new enzymes potentially involved in anaerobic naphthalene degradation by the sulfate-reducing enrichment culture N47. Arch Microbiol 193:241–250
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Christensen N, Batstone DJ, He Z, Angelidaki I, Schmidt JE (2004) Removal of polycyclic aromatic hydrocarbons (PAHs) from sewage sludge by anaerobic degradation. Water Sci Technol 50:237–244
Cox G, Young I, McCann L, Gibson F (1969) Biosynthesis of ubiquinone in Escherichia coli K-12: location of genes affecting the metabolism of 3-octaprenyl-4-hydroxybenzoic acid and 2-octaprenylphenol. J Bacteriol 99:450–458
DiDonato RJ Jr, Young ND, Butler JE, Chin K-J, Hixson KK, Mouser P, Lipton MS, DeBoy R, Methé BA (2010) Genome sequence of the deltaproteobacterial strain NaphS2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS One 5:e14072
Ebenau-Jehle C, Mergelsberg M, Fischer S, Bruls T, Jehmlich N, von Bergen M, Boll M (2017) An unusual strategy for the anoxic biodegradation of phthalate. ISME J 11:224–236
Fuchs G, Boll M, Heider J (2011) Microbial degradation of aromatic compounds—from one strategy to four. Nat Rev Microbiol 9:803–816
Galushko A, Minz D, Schink B, Widdel F (1999) Anaerobic degradation of naphthalene by a pure culture of a novel type of marine sulphate-reducing bacterium. Environ Microbiol 1:415–420
Gerdes K, Møller-Jensen J, Jensen RB (2000) Plasmid and chromosome partitioning: surprises from phylogeny. Mol Microbiol 37:455–466
Griffiths RI, Whiteley AS, O’Donnell AG, Bailey MJ (2000) Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol 66:5488–5491
Gulmezian M, Hyman KR, Marbois BN, Clarke CF, Javor GT (2007) The role of UbiX in Escherichia coli coenzyme Q biosynthesis. Arch Biochem Biophys 467:144–153
Hahne H et al (2013) DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat Methods 10:989–991
Hauck SM et al (2010) Deciphering membrane-associated molecular processes in target tissue of autoimmune uveitis by label-free quantitative mass spectrometry. Mol Cell Proteomics 9:2292–2305
Inoue H, Nojima H, Okayama H (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28
Junghare M, Spiteller D, Schink B (2016) Enzymes involved in the anaerobic degradation of ortho-phthalate by the nitrate-reducing bacterium Azoarcus sp. strain PA01. Environ Microbiol 18:3175–3188
Kleemann R, Meckenstock RU (2011) Anaerobic naphthalene degradation by Gram-positive, iron-reducing bacteria. FEMS Microbiol Ecol 78:488–496
Kung JW et al (2009) Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc Natl Acad Sci USA 106:17687–17692
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lovley DR, Woodward JC, Chapelle FH (1994) Stimulated anoxic biodegradation of aromatic hydrocarbons using Fe(III) ligands. Nature 370:128–131
Lutkenhaus J (2012) The ParA/MinD family puts things in their place. Trends Microbiol 20(9):411–418
Lykidis A et al (2011) Multiple syntrophic interactions in a terephthalate-degrading methanogenic consortium. ISME J 5:122–130
Marozava S, Mouttaki H, Muller H, Laban NA, Probst AJ, Meckenstock RU (2018) Anaerobic degradation of 1-methylnaphthalene by a member of the Thermoanaerobacteraceae contained in an iron-reducing enrichment culture. Biodegradation 29:23–39
Meckenstock RU, Mouttaki H (2011) Anaerobic degradation of non-substituted aromatic hydrocarbons. Curr Opin Biotechnol 22:406–414
Meckenstock RU, Annweiler E, Michaelis W, Richnow HH, Schink B (2000) Anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Appl Environ Microbiol 66:2743–2747
Meckenstock RU et al (2016) Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J Mol Microbiol Biotechnol 26:92–118
Meganathan R (2001) Biosynthesis of menaquinone (vitamin K-2) and ubiquinone (coenzyme Q): a perspective on enzymatic mechanisms. Vitam Horm 61:173–218
Merl J, Ueffing M, Hauck SM, von Toerne C (2012) Direct comparison of MS-based label-free and SILAC quantitative proteome profiling strategies in primary retinal Müller cells. Proteomics 12:1902–1911
Mouttaki H, Johannes J, Meckenstock RU (2012) Identification of naphthalene carboxylase as a prototype for the anaerobic activation of non-substituted aromatic hydrocarbons. Environ Microbiol 14:2770–2774
Musat F et al (2009) Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ Microbiol 11:209–219
Payne KAP et al (2015) New cofactor supports α, β-unsaturated acid decarboxylation via 1,3-dipolar cycloaddition. Nature 522:497–501
Schmitt ME, Brown TA, Trumpower BL (1990) A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res 18:3091–3092
Schühle K, Fuchs G (2004) Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic in phenol metabolism in Thauera aromatica. J Bacteriol 186:4556–4567
Selesi D et al (2010) Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J Bacteriol 192:295–306
White MD et al (2015) UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature 522:502–506
Wu J-H, Wu F-Y, Chuang H-P, Chen W-Y, Huang H-J, Chen S-H, Liu W-T (2013) Community and proteomic analysis of methanogenic consortia degrading terephthalate. Appl Environ Microbiol 79:105–112
Zhang H, Javor GT (2003) Regulation of the isofunctional genes ubiD and ubiX of the ubiquinone biosynthetic pathway of Escherichia coli. FEMS Microbiol Lett 223:67–72
Zhang X, Young LY (1997) Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl Environ Microbiol 63:4759–4764
Acknowledgements
JSK was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Priority Programme 1319 ‘Biological transformations of hydrocarbons without oxygen: from the molecular to the global scale’. The authors declare that there is no conflict of interest. RM acknowledges support by the ERC advanced grant EcOILogy # 666952.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Koelschbach, J.S., Mouttaki, H., Merl-Pham, J. et al. Identification of naphthalene carboxylase subunits of the sulfate-reducing culture N47. Biodegradation 30, 147–160 (2019). https://doi.org/10.1007/s10532-019-09872-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10532-019-09872-z