
Abstract
Background
During chronic lung infections of cystic fibrosis patients Pseudomonas aeruginosa populations undergo extensive evolutionary diversification. However, the selective drivers of this evolutionary process are poorly understood. To test the effects of temperate phages on diversification in P. aeruginosa biofilms we experimentally evolved populations of P. aeruginosa for approximately 240 generations in artificial sputum medium with or without a community of three temperate phages.
Results
Analysis of end-point populations using a suite of phenotypic tests revealed extensive phenotypic diversification within populations, but no significant differences between the populations evolved with or without phages. The most common phenotypic variant observed was loss of all three types of motility (swimming, swarming and twitching) and resistance to all three phages. Despite the absence of selective pressure, some members of the population evolved antibiotic resistance. The frequency of antibiotic resistant isolates varied according to population and the antibiotic tested. However, resistance to ceftazidime and tazobactam-piperacillin was observed more frequently than resistance to other antibiotics, and was associated with higher prevelence of isolates exhibiting a hypermutable phenotype and increased beta-lactamase production.
Conclusions
We observed considerable within-population phenotypic diversity in P. aeruginosa populations evolving in the artificial sputum medium biofilm model. Replicate populations evolved both in the presence and absence of phages converged upon similar sets of phenotypes. The evolved phenotypes, including antimicrobial resistance, were similar to those observed amongst clinical isolates from cystic fibrosis infections.
Similar content being viewed by others


Background
Pseudomonas aeruginosa is an important opportunistic pathogen that can cause chronic lung infections in patients with cystic fibrosis (CF) [1] or non-CF bronchiectasis [2]. Once established as a chronic infection, a P. aeruginosa strain can remain in the lungs of a patient for life, despite frequent use of antibiotic therapy. During the infection, the pathogen adopts a biofilm lifestyle, with evidence from explanted lungs suggesting the formation of free aggregates rather than surface-attached biofilms [3]. Throughout these long time periods, populations of P. aeruginosa adapt to the CF lung in several ways, with isolates often exhibiting phenotypes such as mucoidy, auxotrophy, hypermutability, loss of known virulence factors, loss of motility, slower growth in rich media and resistance to antimicrobials [4–7]. In addition to flagella-mediated swimming motility, P. aeruginosa exhibits motility on solid or semi-solid surfaces, using IV pili for twitching motility [8], and both flagella and type IV pili for swarming motility [9]. In CF, mutations occur in genes encoding both type IV pili and flagella.
Recently, it has also become evident that P. aeruginosa populations in the CF lung can diversify both phenotypically [7, 10–16] and genetically [14, 17]. In particular, we have shown that populations of the most common clone of P. aeruginosa infecting CF patients in the UK, the Liverpool Epidemic Strain (LES) [18, 19], are highly diverse [11, 12, 20]. However, the factors driving and maintaining this diversity, also observed in other CF pathogens [21], are poorly understood.
It has been shown that sub-inhibitory concentrations of antibiotics can drive diversification of P. aeruginosa populations [22], but other possible contributory factors include host-responses, other members of the complex microbial community associated with the CF lung [23] or the biofilm lifestyle itself [24]. Temperate phages are abundant in the CF lung [25], and we have demonstrated previously that LES temperate phages can both mediate and drive adaptive evolution during experimental evolution in a biofilm model system, by increasing the supply of mutations in genes involved in type IV pilin biogenesis and quorum sensing through insertional inactivation of bacterial genes [26, 27]). The LES phages have also been implicated in the competitiveness of the strain in chronic infection models [28, 29].
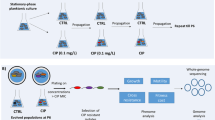
To test the hypothesis that temperate phages play an important role in driving phenotypic diversity, here we analysed a suite of phenotypic traits in populations of P. aeruginosa that have been experimentally evolved for approximately 240 bacterial generations with or without an assemblage of three LES temperate phages (LESφ2, φ3 and φ4) in Artificial Sputum Medium (ASM). ASM is an in vitro environment that recapitulates the physiochemical properties of lung sputum and in which P. aeruginosa forms free-floating biofilms [30] and expresses biofilm-associated genes [31].
Results
Phenotypic diversification and loss of wild-type phenotypes occurred in both treatment and control populations
Twelve replicate populations of P. aeruginosa PAO1 were experimentally evolved for ~240 generations in ASM in the absence (#1-6) [control treatment] or presence (#7-12) of three temperate LES phages. At the end of the evolution experiment, 40 randomly-selected isolates per population were characterised by assessing a suite of phenotypic traits previously associated with adaptation to the CF lung [7]. These were three types of motility (swimming, swarming and twitching), auxotrophy, hypermutability, and resistance to each of the three LES phages (LESφ2, φ3 and φ4). These data are available in Additional File 1. Extensive phenotypic diversity was observed in all endpoint populations. Analysis of molecular variance (AMOVA) indicated that the majority of the phenotypic variance was associated with diversity within (74%) and between populations (24%), whereas less than 2% of the variation was explained by treatment (i.e. presence/absence of phages) (Table 1). From the 480 isolates, 57 different haplotypes were identified, based on eight phenotypic traits. The haplotype of the ancestor was very rare in the evolved populations; only 8/480 isolates shared the ancestral haplotype, all of which were recovered from the control treatment. The most common haplotype constituted 23% of isolates and was observed in all but one population. This haplotype was characterised by loss of all three types of motility and resistance to all three LES phages, but displayed the wild-type mutation rate and growth on minimal media phenotypes.
In order to visualise variation between populations, a principal component analysis (PCA) was applied to the multivariate phenotypic dataset (Fig. 1). The first principal component was strongly associated with twitching motility (Fig. 1a). There was a large degree of overlap between isolates from the two treatments, suggesting that populations from the different treatments evolved along similar trajectories (Fig. 1b). While some degree of clustering by population was observed, extensive diversification within individual populations was evident (Fig. 1c). Many isolates exhibited loss of multiple phenotypes, but there was considerable variation in the proportion of mutants in each population (Fig. 2). However, there was no difference between the control and phage treatments for any of the phenotypes (Wilcoxon rank-sum test, P > 0.05).

Exploratory principal component analysis of multivariate phenotype data. (a) Variable factor map, (b) individual factor map, labelled with experimental treatment and (c) individual factor map, labelled with population number. The Eigenvalues for the first and second components were 2.07 and 1.59, respectively

Boxplots of the proportion of isolates per population displaying the mutant phenotype for twitching motility (a), swarming motility (b), swimming motility (c), hypermutability (d) and auxotrophy (e). Thick black horizontal lines represent the median, the box represents the upper and lower quantiles, and the circles represent outliers. 40 isolates were tested per populations (6 populations per treatment)
Endpoint isolates were tested for resistance to each of the LES phages. Phage resistance was widespread in both treatments (Fig. 3). The majority of isolates were resistant to all three phages in all but one of the populations that was not exposed to phages (#5), in which 93% of isolates remained susceptible to all three phages.

Phage resistance of endpoint isolates to LESφ2-4. Bars are coloured according to the particular phage combination to which resistance was observed. High titre pure phage stocks were spotted onto a soft-agar lawn of each isolate and scored as resistant if no lysis was observed after overnight incubation at 37 °C
Resistance to antibiotics
Disk diffusion assays revealed that all of the isolates tested (40/40) from every population were as susceptible to a panel of antibiotics as the ancestor. The method was therefore adapted to look for resistant isolates that may be present at a low frequency in the population. Entire populations were spread on to agar plates containing antibiotics at clinically relevant resistance breakpoint levels. The frequency of antibiotic resistant isolates varied according to the population and the antibiotic (Fig. 4). The highest frequency of tazobactam-piperacillin resistance, observed in the phage-treated population #11 (0.08%), exceeded that seen with the control mutator strain, PAO1ΔmutS. Resistance to ceftazidime and tazobactam-piperacillin arose more frequently than resistance to other antibiotics. No meropenem resistant isolates were identified.

Frequency of bacterial cells in whole populations displaying clinical resistance to a panel of antibiotics (arcsine transformed). Frequencies were calculated after plating endpoint populations onto antibiotic free media, and media containing antibiotics at the clinical resistance breakpoint level. Planktonic PAO1 and PAO1 grown in ASM were included as negative controls. PAO1ΔmutS was included as a mutator strain control. Error bars ±1 S.E.M. Piperacillin/tazobactam (TZP); ceftazidime (CAZ); ciprofloxacin (CIP); tobramycin (TOB); colistin (COL)
In order to assess the extent to which the mutator phenotype was linked to the high frequencies of antibiotic resistant cells, the frequency of hypermutators in a population was plotted against the frequency of isolates resistant to ceftazidime and tazobactam-piperacillin (Fig. 5). Higher frequencies of hypermutators were linked to frequencies of resistance against both tazobactam-piperacillin (Spearman’s rank correlation coefficient: ρ = 0.69, p < 0.05), and ceftazidime (Spearman’s rank correlation coefficient: ρ = 0.72, p < 0.01). To further characterise the nature of the spontaneously occurring antibiotic resistance, Minimum Inhibitory Concentration (MIC) values were determined for two sets of 20 colonies from population #11 identified as resistant to either ceftazidime or tazobactam-piperacillin. The MIC values for ceftazidime (32–128 mg l−1); and tazobactam-pipericillin (128–256 mg l−1) were at least two-fold higher than the clinical resistance breakpoints (8 mg l−1 and 16 mg l−1 respectively [32]). All isolates found to be resistant to one antibiotic were also resistant to the other. A chromogenic beta-lactamase detection assay was conducted on five randomly selected isolates from each antibiotic medium, using nitrocefin. Beta-lactamase production above the level of the control (PAO1) was observed in all of the isolates tested (Fig. 6). Antibiotic resistance data and data for beta-lactamase production are available in Additional File 2.

The correlation between the frequency of hypermutators in a population and the frequency of resistant isolates (arcsine transformed). Ceftazidime (CAZ); piperacillin/tazobactam (TZP); n = 12

Beta-lactamase production in ceftazidime and tazobactam-piperacillin resistant isolates. Activity was measured using nitrocefin, in a chromogenic assay by calculating the change in A486/390 per minute. The dashed line indicates the value of the negative control (PAO1). Ceftazidime (CAZ); piperacillin/tazobactam (TZP). Bars indicate standard error
Discussion
Consistent with clinical studies reporting extensive phenotypic diversity in P. aeruginosa populations recovered from the sputa of CF patients [10, 11, 13, 14], populations of P. aeruginosa grown in ASM exhibited substantial phenotypic heterogeneity. Notably we observed the evolution of many phenotypic variants that are also commonly found amongst isolates from CF patients. As with naturally occurring populations in the CF lung [10, 11], the majority of the diversity in our evolved populations was within populations rather than between populations. Although our previous genomic analyses suggested that the phages play a key role in accelerating the evolution of key phenotypes [26], our populations treated with or without phages evolved along similar trajectories, and converged upon similar suites of evolved phenotypes [26, 33].
Previous studies of CF isolates have reported loss of swimming, swarming and twitching motility [10]. Although it has been proposed that this loss of motility may be an immune avoidance strategy [4], the lack of any immune system in our model suggests that loss of motility may evolve, at least in part, because of other selective pressures. These could include the viscosity of the lung environment, though this remains a hypothesis yet to be tested. Although a role for type IV pili in biofilm formation has been suggested [34, 35], a study examining clinical isolates observed no correlation between biofilm production and motility [36]. Our observations suggest that type IV pili are not required for the formation of the kind of free-floating biofilms that occur in the ASM model.
A high frequency of hypermutators was observed in some evolved populations, but there were large differences between populations, ranging from 0 to > 90% hypermutators. An elevated mutation rate can be detrimental due to the increased risk of deleterious mutations, but hypermutability can reach fixation in a population by hitch-hiking with beneficial mutations [37], and can be advantageous in a novel environment, accelerating adaptation [38]. The mutator phenotype has been shown to occur during biofilm growth and may confer intrinsic fitness benefits, such as significantly enhanced microcolony growth [39], or overproduction of the H2O2-degrading enzyme catalase, leading to increased resistance to oxidative stress [40]. The hypermutability phentotype has also been linked with antibiotic resistance [41, 42], and in our study a clear correlation was observed between the frequency of hypermutators in a population and the frequency of clinically resistant isolates. Increased basal production of Beta-lactamase has been observed previously in mutator strains [43]. The finding that clinically resistant isolates are present at low frequency in hypermutable populations, even in the absence of antibiotic selection, suggests that factors other than antibiotic therapy per se can drive antimicrobial resistance in chronic lung infections. The role of mutator phenotypes in driving evolution in P. aeruginosa surface-attached biofilm systems, including an increase in the frequency of mutations implicated in antibiotic resistance, has been described previously [39]. Evolution of antibiotic resistance has also been shown during biofilm growth for Escherichia coli [44]. Here, we demonstrate the development of resistance in a free-floating biofilm model systems designed to mimic sputum.
The diversification observed in ASM over this relatively short timescale could be driven by growth in a biofilm. It is known that spatially structured environments, like the viscous ASM, can select for the evolution of diversity [45]. Previous studies further suggest that P. aeruginosa diversifies when grown as a surface-attached biofilm, and this diversity helps protect against oxidative stress [24]. It has been suggested that this was due to the “insurance effect”, which posits that biodiversity has a buffering effect over time on productivity, as well as raising the overall mean productivity of an ecosystem [46]. However, the applicability of the insurance hypothesis to bacterial biofilms has been questioned [47]. Our observations suggest that diversification of biofilm populations, especially those that have evolved hypermutability, could potentially increase their evolutionary responsiveness in the event of antibiotic treatment by providing standing genetic variation for antibiotic resistance upon which natural selection can act.
Conclusions
We have demonstrated that many of the phenotypic adaptations observed amongst P. aeruginosa isolates from CF chronic infections occur during evolution in a model designed to mimic the CF sputum environment, even in the absence of any selection due to host responses or antibiotics. Although we have shown previously that phages can alter the mode and rate of adaptive evolution [26], comparable sets of phenotypes eventually occurred both in the presence and absence of phages. It is, however, worth noting that similar phenotypes can be driven by sub-inhibitory concentrations of antibiotics in non-biofilm systems [48]. In CF, it has been proposed that the maintenance of phenotypically diverse P. aeruginosa populations may be a consequence of the spatial heterogeneity of the lung, leading to regional isolation of separately evolving communities [15]. Here we show that similar population heterogeneity occurs in an artificial sputum biofilm system, emphasizing the potential utility of this model as a tool to facilitate a better understanding of the factors driving P. aeruginosa adaptation and diversification during infections.
Methods
Bacterial strains, bacteriophages and growth conditions
The laboratory reference strain P. aeruginosa PAO1, originally isolated from a wound in Melbourne, Australia in 1954 [49], was used as a model host because it is known to be fully susceptible to LES φ2-4. P. aeruginosa LESB58 was isolated from a CF patient in Liverpool in 1988 [19]. Bacteriophages were isolated from the P. aeruginosa LESB58 as described previously [50]. The challenge experiment was performed using PAO1 cultures grown in ASM [22, 30] at 37 °C, 60 rpm for 96 h periods as described previously [26]. Prior to the challenge experiment and in follow up experiments, all bacterial strains and phages were grown and propagated in standard Luria Bertani broth (LB). Phage suspensions (1 × 108 – 1 × 1011 p.f.u ml−1) were stored in LB at 4 °C.
Challenge experiment
Twelve replicate ASM cultures (hereafter referred to as populations) were initiated by inoculation of approximately 5 × 107 cells of mid-exponential phase PAO1 into 5 ml ASM, followed by 24 h incubation (37 °C, 60 rpm). Equal numbers of each LES phage (LES φ2 – 4) were then added, once only, to 6 replicates (populations 7–12), to a total multiplicity of infection (MOI) of 0.1. The remaining six replicates were designated phage-free controls (populations 1–6). Each population was incubated for a further 72 h before degradation of biofilm structures using Sputasol (Oxoid) as described previously [30]. The biofilm homogenate was transferred (1:100) into fresh ASM (12 × 5 ml) and incubated for a further 96 h. This process was repeated every 96 h for 120 days (30 transfers and approximately 240 bacterial generations).
Phenotypic diversity of endpoint populations
At the end of the challenge experiment, 40 evolved isolates were obtained from each population by plating onto Columbia media and patching of randomly selected colonies onto fresh Columbia media. All isolates were stored in LB with 30% (v/v) glycerol at −80 °C. A total of 240 isolates for each treatment were subjected to a series of phenotypic tests.
For growth rate in LB, overnight cultures were diluted 1:100 and 200 μl added to wells (n = 4 per isolate) of a clear-bottomed 96 well plate. The plate was held at 37 °C, with shaking at 100 r.p.m. in an Omega fluostar plate reader. The OD600 was measured every 5 min and the doubling time in the exponential phase of growth calculated for each well. Two biological replicates were performed per isolate. Ten isolates were tested per population.
For auxotrophy, a single colony was patched onto M9 minimal media and incubated at 37 °C. Isolates displaying no growth after 18 h were classed as auxotrophic. For hypermutability, 10-fold serially diluted overnight cultures grown in LB were spotted onto LB agar plates containing 300 μg ml−1 rifampicin. Plates were incubated overnight at 37 °C. As described previously [12], isolates were classed as hypermutable if growth on rifampicin was comparable to PAOΔmutS (ie. different by less than 10-fold), a known hypermutator.
For swarming motility, a small portion of a colony was lightly touched on to the surface of media containing 0.5% (w/v) bacteriological agar as described previously [51]. Plates were incubated for 16 h at 37 °C and the diameter of the swarm was measured at the widest point, using a strain PAO1 pilA mutant as a negative control. An isolate with a diameter <6 mm was considered swarming deficient. For swimming motility, a small portion of a colony was lightly touched to the surface of a swimming plate [52]. Plates were wrapped loosely in clingfilm to prevent dehydration, and incubated without inversion for 14 h. The visible diameter of bacterial growth was measured at the widest point. Using the non-motile P. aeruginosa LESB58 was used as a negative control, an isolate with a diameter <20 mm was considered swimming deficient. Twitching motility was measured in duplicate as described previously [53], using ancestral PAO1 as a positive control. Isolates with a zone diameter <10 mm were considered to have impaired twitching motility.
To test bacterial resistance to the phages used in this study (LESφ2, φ3 and φ4), a modified version of the phage spot assay was used. 20 μl of 10-fold diluted overnight culture was mixed with 200 μl agar overlay and poured into a well containing 1 ml bottom agar, in a 25 well square petri dish (Fisher Scientific). High titre (>1x1010 pf.u. ml-1) preparations of each phage were spotted on top. Isolates were classed as resistant to a phage if there was no evidence of bacterial lysis after overnight incubation at 37 °C.
Antibiotic susceptibilities
Susceptibilities to six antibiotics were determined using the disc diffusion method, as described previously [11]. The antibiotics were: piperacillin/tazobactam, ceftazidime, meropenem, ciprofloxacin, tobramycin and colistin. To detect resistant isolates at a low abundance in a population, a modified version of the agar dilution method was developed. Whole population cultures were adjusted to the turbidity equivalence of a McFarland standard 0.5 (Beckton Dickinson), and 100 μl was plated directly onto Mueller-Hinton Agar plates containing antibiotics at the breakpoint levels for clinical resistance [32]. After overnight growth at 37 °C, colonies growing on antibiotic-containing media were calculated as a proportion of total population counts. “Planktonic” PAO1 grown in LB and PAO1 grown in ASM were included as controls.
Beta-lactamase assay of beta-lactam resistant isolates
Isolates were passaged twice through antibiotic-free media (LB agar) to ensure basal (as opposed to induced) beta-lactamase levels were being studied. Cultures were grown in LB to an Absorbance at 600 nm of 0.3, and nitrocefin (Merck-Millipore) was added to a final concentration of 51.6 μg ml−1. As the λmax of nitrocefin changes from 390 nm (in the absence of beta-lactamase) to 486 nm upon hydrolysis, the absorbance at both wavelengths was measured every 5 min, for 35 min. The A486 nm/390 nm ratio was calculated for every time point as an indicator of beta-lactamase production. Five biological replicates were performed for each isolate.
Analysis of molecular variance (AMOVA)
At the end of the challenge experiment, 40 evolved isolates were selected at random from each population and screened for eight phenotypic traits (the three types of motility, auxotrophy, hypermutability, resistance to each of the three phages). Isolates were classified as different haplotypes based on the combination of phenotypic traits displayed. For each trait, isolates were scored a 0 if they displayed the same phenotype as the ancestor, or a 1 if they displayed a mutant phenotype. Here we define a haplotype as a unique combination of phenotypic traits. The evolutionary distance between the haplotypes was estimated by creation of a Euclidean distance matrix. AMOVA was performed using the ade4 package in R [54]; the distance matrix was partitioned into sub-matrices for the various subdivisions of the data, including treatment (control or phage-treated), population and individual isolates. The sums of squares were computed and analysed in a nested ANOVA framework and significance tested by Monte-Carlo permutation tests (49 permutations). Such an approach has been used previously to assess diversity of clinical P. aeruginosa isolates, using phenotype data [11].
Principal component analysis (PCA)
Phenotypic data from the host-phage coevolution experiment resulted in large, multivariate datasets. To explore the underlying structure of the data and elucidate the factors that are responsible for much of the variance, PCA was used as an exploratory data analysis tool, using the ade4 package [54] in R. Complete datasets (i.e. data for all 40 isolates within each population) were available for all but two of the variables (biofilm formation and doubling time in LB). For the incomplete variables, only the first 10 isolates of every population were tested, due to the large amount of time and work required to test these specific phenotypes. PCA in R cannot proceed with missing values, but numerous methods exist to allow missing values to be ignored or replaced. Multiple imputation was chosen as a method to replace the missing data, using the mi package [55] in R, as it enables existing data to be retained. Data were plotted with dudi PCA scatter plots, and the independent variables (phage-treatment and population) overlaid, using the s.class function. Data for seven phenotypes were included in the PCA analysis: swimming, swarming and twitching motility, hypermutability, growth rate, biofilm formation and auxotrophy.
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- ANOVA:
-
Analysis of variance
- ASM:
-
Artificial sputum medium
- CF:
-
Cystic fibrosis
- LB:
-
Luria Bertani
- LES:
-
Liverpool Epidemic Strain
- PCA:
-
Principal component analysis
References
Hoiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 2011;9:32.
McDonnell MJ, Jary HR, Perry A, MacFarlane JG, Hester KL, Small T, Molyneux C, Perry JD, Walton KE, De Soyza A. Non cystic fibrosis bronchiectasis: a longitudinal retrospective observational cohort study of Pseudomonas persistence and resistance. Respir Med. 2015;109(6):716–26.
Bjarnsholt T, Jensen PO, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Hoiby N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol. 2009;44(6):547–58.
Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. 1994;62(2):596–605.
Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D’Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci. 2006;103(22):8487–92.
Ciofu O, Mandsberg LF, Bjarnsholt T, Wassermann T, Høiby N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology. 2010;156(4):1108–19.
Winstanley C, O’Brien S, Brockhurst MA. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 2016;24:327–37.
Burrows LL. Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu Rev Microbiol. 2012;66:493–520.
Overhage J, Bains M, Brazas MD, Hancock RE. Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J Bacteriol. 2008;190(8):2671–9.
Workentine ML, Sibley CD, Glezerson B, Purighalla S, Norgaard-Gron JC, Parkins MD, Rabin HR, Surette MG. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One. 2013;8(4):e60225.
Mowat E, Paterson S, Fothergill JL, Wright EA, Ledson MJ, Walshaw MJ, Brockhurst MA, Winstanley C. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Respir Crit Care Med. 2011;183(12):1674–9.
Fothergill JL, Mowat E, Ledson MJ, Walshaw MJ, Winstanley C. Fluctuations in phenotypes and genotypes within populations of Pseudomonas aeruginosa in the cystic fibrosis lung during pulmonary exacerbations. J Med Microbiol. 2010;59:472–81.
Ashish A, Paterson S, Mowat E, Fothergill JL, Walshaw MJ, Winstanley C. Extensive diversification is a common feature of Pseudomonas aeruginosa populations during respiratory infections in cystic fibrosis. J Cyst Fibros. 2013;12(6):790–3.
Darch SE, McNally A, Harrison F, Corander J, Barr HL, Paszkiewicz K, Holden S, Fogarty A, Crusz SA, Diggle SP. Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci Rep. 2015;5:7649.
Jorth P, Staudinger BJ, Wu X, Hisert KB, Hayden H, Garudathri J, Harding CL, Radey MC, Rezayat A, Bautista G, et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe. 2015;18(3):307–19.
Clark ST, Diaz Caballero J, Cheang M, Coburn B, Wang PW, Donaldson SL, Zhang Y, Liu M, Keshavjee S, Yau YC, et al. Phenotypic diversity within a Pseudomonas aeruginosa population infecting an adult with cystic fibrosis. Sci Rep. 2015;5:10932.
Williams D, Evans B, Haldenby S, Walshaw MJ, Brockhurst MA, Winstanley C, Paterson S. Divergent, coexisting Pseudomonas aeruginosa lineages in chronic cystic fibrosis lung infections. Am J Respir Crit Care Med. 2015;191(7):775–85.
Martin K, Baddal B, Mustafa N, Perry C, Underwood A, Constantidou C, Loman N, Kenna DT, Turton JF. Clusters of genetically similar isolates of Pseudomonas aeruginosa from multiple hospitals in the UK. J Med Microbiol. 2013;62(Pt 7):988–1000.
Winstanley C, Langille MG, Fothergill JL, Kukavica-Ibrulj I, Paradis-Bleau C, Sanschagrin F, Thomson NR, Winsor GL, Quail MA, Lennard N, et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 2009;19(1):12–23.
Williams D, Paterson S, Brockhurst MA, Winstanley C: Refined analyses suggest that recombination is a minor source of genomic diversity in Pseudomonas aeruginosa chronic cystic fibrosis infections. Microb Genom 2016. doi: 10.1099/mgen.0.000051.
Lieberman TD, Michel JB, Aingaran M, Potter-Bynoe G, Roux D, Davis Jr MR, Skurnik D, Leiby N, LiPuma JJ, Goldberg JB, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43(12):1275–80.
Wright EA, Fothergill JL, Paterson S, Brockhurst MA, Winstanley C. Sub-inhibitory concentrations of some antibiotics can drive diversification of Pseudomonas aeruginosa populations in artificial sputum medium. BMC Microbiol. 2013;13:170.
Rabin HR, Surette MG. The cystic fibrosis airway microbiome. Curr Opin Pulm Med. 2012;18(6):622–7.
Boles BR, Thoendel M, Singh PK. Self-generated diversity produces “insurance effects” in biofilm communities. Proc Natl Acad Sci U S A. 2004;101(47):16630–5.
James CE, Davies EV, Fothergill JL, Walshaw MJ, Beale CM, Brockhurst MA, Winstanley C. Lytic activity by temperate phages of Pseudomonas aeruginosa in long-term cystic fibrosis chronic lung infections. ISME J. 2015;9(6):1391–8.
Davies E, James CE, Williams D, O’Brien S, Fothergill JL, Haldenby S, Paterson S, Winstanley C, Brockhurst MA. Temperate phages both mediate and drive adaptative evolution in pathogen biofilms. Proc Natl Acad Sci U S A. 2016;113:8266–71.
Burns N, James CE, Harrison E. Polylysogeny magnifies competitiveness of a bacterial pathogen in vivo. Evol Appl. 2015;8(4):346–51.
Lemieux AA, Jeukens J, Kukavica-Ibrulj I, Fothergill JL, Boyle B, Laroche J, Tucker NP, Winstanley C, Levesque RC. Genes required for free phage production are essential for pseudomonas aeruginosa chronic lung infections. J Infect Dis. 2016;213(3):395–402.
Davies EV, James CE, Kukavica-Ibrulj I, Levesque RC, Brockhurst MA, Winstanley C. Temperate phages enhance pathogen fitness in chronic lung infection. ISME J. 2016;10(10):2553–5.
Kirchner S, Fothergill JL, Wright EA, James CE, Mowat E, Winstanley C. Use of artificial sputum medium to test antibiotic efficacy against Pseudomonas aeruginosa in conditions more relevant to the cystic fibrosis lung. J Vis Exp. 2012;64:e3857.
Fothergill JL, Neill DR, Loman N, Winstanley C, Kadioglu A. Pseudomonas aeruginosa adaptation in the nasopharyngeal reservoir leads to migration and persistence in the lungs. Nat Commun. 2014;5:4780.
The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. 2016. http://www.eucast.org, Version 6.0.
Cazares A, Mendoza-Hernandez G, Guarneros G. Core and accessory genome architecture in a group of Pseudomonas aeruginosa Mu-like phages. BMC Genomics. 2014;15:1146.
Klausen M, Heydorn A, Ragas P, Lambertsen L, Aaes-Jorgensen A, Molin S, Tolker-Nielsen T. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol Microbiol. 2003;48(6):1511–24.
O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30(2):295–304.
Head NE, Yu H. Cross-sectional analysis of clinical and environmental isolates of Pseudomonas aeruginosa: biofilm formation, virulence, and genome diversity. Infect Immun. 2004;72(1):133–44.
Chao L, Cox EC: Competition between high and low mutating strains of Escherichia coli. Evolution. 1983;37:125–34.
Sniegowski PD, Gerrish PJ, Lenski RE. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387(6634):703–5.
Conibear TC, Collins SL, Webb JS. Role of mutation in Pseudomonas aeruginosa biofilm development. PLoS One. 2009;4(7):e6289.
Torres-Barceló C, Cabot G, Oliver A, Buckling A, MacLean RC. A trade-off between oxidative stress resistance and DNA repair plays a role in the evolution of elevated mutation rates in bacteria. Proc R Soc B Biol Sci. 2013;280(1757):20130007.
Waine DJ, Honeybourne D, Smith EG, Whitehouse JL, Dowson CG. Association between hypermutator phenotype, clinical variables, mucoid phenotype, and antimicrobial resistance in Pseudomonas aeruginosa. J Clin Microbiol. 2008;46(10):3491–3.
Plasencia V, Borrell N, Macía MD, Moya B, Pérez JL, Oliver A. Influence of high mutation rates on the mechanisms and dynamics of in vitro and in vivo resistance development to single or combined antipseudomonal agents. Antimicrob Agents Chemother. 2007;51(7):2574–81.
Mandsberg LF, Ciofu O, Kirkby N, Christiansen LE, Poulsen HE, Høiby N. Antibiotic resistance in Pseudomonas aeruginosa strains with increased mutation frequency due to inactivation of the DNA oxidative repair system. Antimicrob Agents Chemother. 2009;53(6):2483–91.
Tyerman JG, Ponciano JM, Joyce P, Forney LJ, Harmon LJ. The evolution of antibiotic susceptibility and resistance during the formation of Escherichia coli biofilms in the absence of antibiotics. BMC Evol Biol. 2013;13:22.
Habets MG, Rozen DE, Hoekstra RF, de Visser JA. The effect of population structure on the adaptive radiation of microbial populations evolving in spatially structured environments. Ecol Lett. 2006;9(9):1041–8.
Yachi S, Loreau M. Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis. Proc Natl Acad Sci. 1999;96(4):1463–8.
Cooper T, Beaumont H, Rainey P. Biofilm diversity as a test of the insurance hypothesis. Microbiology. 2005;151(9):2815–6.
Wassermann T, Meinike Jorgensen K, Ivanyshyn K, Bjarnsholt T, Khademi SM, Jelsbak L, Hoiby N, Ciofu O. The phenotypic evolution of Pseudomonas aeruginosa populations changes in the presence of subinhibitory concentrations of ciprofloxacin. Microbiology. 2016;162(5):865–75.
Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406(6799):959–64.
James C, Fothergill J, Kalwij H, Hall A, Cottell J, Brockhurst M, Winstanley C. Differential infection properties of three inducible prophages from an epidemic strain of Pseudomonas aeruginosa. BMC Microbiol. 2012;12(1):216.
Tremblay J, Deziel E. Improving the reproducibility of Pseudomonas aeruginosa swarming motility assays. J Basic Microbiol. 2008;48(6):509–15.
Deziel E, Comeau Y, Villemur R. Initiation of biofilm formation by Pseudomonas aeruginosa 57RP correlates with emergence of hyperpiliated and highly adherent phenotypic variants deficient in swimming, swarming, and twitching motilities. J Bacteriol. 2001;183(4):1195–204.
Häußler S, Ziegler I, Löttel A, Götz FV, Rohde M, Wehmhöhner D, Saravanamuthu S, Tümmler B, Steinmetz I. Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J Med Microbiol. 2003;52(4):295–301.
Dray ABDS. The ade4 package:implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20.
Su YS, Gelman A, Hill J, Yajima M. Multiple imputation with diagnostics (mi) in R: Opening windows into the black box. J Stat Softw. 2011;45(2):1–31.
Acknowledgements
This work was funded by a project grants from The Wellcome Trust (089215/Z/09/Z awarded to CW & MAB) and a studentship co-funded by Medical Research Council and the Institute of Infection and Global Health, University of Liverpool.
Availability of data and materials
No data requiring deposition was generated. All data are included in Additional Files 1 and 2.
Authors’ contributions
ED and CJ contributed equally and carried out all of the experimental work, helped draft the manuscript and analysed data. MB and CW contributed equally, jointly conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
No ethical approval was required.
Author information
Authors and Affiliations
Corresponding author
Additional information
Emily V. Davies and Chloe E. James are equal first authors.
Michael A. Brockhurst and Craig Winstanley are equal senior authors.
Additional files
Additional file 1:
Phenotypic data from this study. (XLSX 139 kb)
Additional file 2:
Antimicrbial susceptibility data and data for beta-lactamase production. (XLSX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Davies, E.V., James, C.E., Brockhurst, M.A. et al. Evolutionary diversification of Pseudomonas aeruginosa in an artificial sputum model. BMC Microbiol 17, 3 (2017). https://doi.org/10.1186/s12866-016-0916-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-016-0916-z